

Abstract
Genetic transformation of plants often results in multiple copies of the introduced DNA at a single locus. To ensure that only a single copy of a foreign gene resides in the plant genome, we used a strategy based on site-specific recombination. The transformation vector consists of a transgene flanked by recombination sites in an inverted orientation. Regardless of the number of copies integrated between the outermost transgenes, recombination between the outermost sites resolves the integrated molecules into a single copy. An example of this strategy has been demonstrated with wheat transformation, where four of four multiple-copy loci were resolved successfully into single-copy transgenes.
Keywords: wheat transformation, site-specific recombination, Cre-lox
The production of transgenic crop plants is an expanding component of agricultural biotechnology. For commercial success, it will be crucial that the introduced traits be transmitted faithfully through successive generations in a predictable manner. Unfortunately, this does not always hold true, as transgene inactivation is becoming a frequently observed phenomenon. Though not well understood, certain factors affecting transgene inactivation have been described that include multiple-copy integration (1–3), differential base composition between transgene and the integration site (4), hyperexpression of transgenes (5, 6), and environmental factors (7). Although single-copy transgenes can be silenced (5, 7), there is a much higher incidence of gene instability correlated with high transgene copy number (1–3, 8). Complex integration sites undergo either structural instability such as intrachromosomal recombination between multiple copies, sometimes resulting in loss of the transgene (1, 9, 10), or chemical modification such as DNA methylation (11).
Potential means to ensure the stability of a transgene include insulating the transgene with matrix-attachment regions (4), avoiding repetition of promoter or transgene sequences, particularly inverted repeats (12), and using moderate promoters to avoid hyperexpression (8, 13). For proper implementation of these features, it would be critical to start with a single, intact copy of the DNA. The conventional method to achieve this aim is to screen among the pool of transformants (surveyed in ref. 8). However, the occurrence of plants containing a single, intact transgene in cereals is rare (14, 15). In the past 7 years, there have been more than 14 independent reports of wheat transformation (15–27). Based on DNA hybridization data of the analyzed plants, less than 10 are potential single-copy integrants (8 of 85 biolistic transformants and ≈1–2 of 26 Agrobacterium-mediated transformants). Because of low transformation efficiencies, obtaining transgenic wheat is, by itself, a challenging task. If the goal is to achieve single-copy lines, then the task becomes an order of magnitude greater.
Different approaches have been used to enrich for single-copy transgenic lines. An “Agrolistic” method has been described based on cobombarding VirD genes along with T-DNA borders flanking the introduced transgene. The resulting transgenic maize calli were found to contain one to two transgene copies at 10–35% efficiency. However, the cointegration of VirD DNA is a frequent event (28, 29). In another approach, the treatment with niacinamide, which is believed to be an inhibitor of recombination, yielded single-copy wheat lines at ≈8% efficiency (15). To achieve near-100% recovery of single-copy lines, we designed a strategy in which every multicopy locus can be converted to a single-copy state. Concomitant with this conversion is the removal of the marker gene. Selectable markers are not necessary after transformation, and their presence precludes using the same selection scheme for subsequent transformations (30, 31). Moreover, one preliminary report attributed a reduction in agronomic performance to the presence of the selectable marker.¶ In this report, we describe generating single-copy, marker-free transgenic wheat lines from each of four different complex-integration loci.
MATERIALS AND METHODS
Transformation Vectors.
Plasmids were constructed by standard recombinant DNA methods and contain pUC18 backbones. The Cre-lox system consists of the 38-kDa Cre recombinase that recognizes a 34-bp target sequence known as lox (32, 33). Various lox alleles have been described (34, 35). The 7-kb plasmid pVS11 (Fig. 1A) contains a fragment flanked by synthetic lox511 alleles of opposite orientation in pUC18. Within this fragment lies a bar-coding region flanked by loxP sites in direct orientation. Upstream and downstream of bar are, respectively, the rice actin promoter (36), denoted as P1, and the maize ubiquitin promoter (37), denoted as P2. Although not relevant to this report, an FRT site (FLP-FRT recombination system) is inserted upstream of P1 (not shown in the figures), intended for use in future recombination experiments. Plasmids pP2-cre (6.4 kb) and pP2-bar (5.5 kb) are also described in Fig. 1A.
Figure 1.

(A) Schematic map (not to scale) of transformation vectors pVS11, pP2-bar, and pP2-cre. Only relevant features and DNA segments are shown. Solid arrowhead, loxP; open arrowhead, lox511. P1, rice actin promoter; P2, maize ubiquitin promoter; bar, phosphinothricin acetyl transferase coding region; cre, Cre recombinase coding region; E, EcoRI; H, HindIII; P, PstI; X, XhoI. Restriction fragments that hybridize to the probes (a, b, or c) are in kilobases. Transgene–host DNA junction fragments that hybridize to probe b are indicated by dotted arrows. Not shown are the pUC18 vector backbones and the nos3′ (transcription terminator) fragments that lie immediately downstream of bar and cre. (B) Resolution product from recombination of lox sites. pVS11 may integrate in multiple copies, with the outermost copies forming four possible configurations. Recombination between the outermost lox511 sites (indicated by dashed lines) resolves the complex locus into a unit copy of pVS11. Recombination between loxP sites removes the selectable marker.
Wheat Transformation.
Immature embryos, collected from greenhouse or field-grown Spring wheat (var. Bobwhite), were transformed by particle bombardment (38) with pVS11 or by equal amounts of pP2-cre and pP2-bar. Bombarded embryos were cultured and selected in the presence of bialaphos (3 mg/liter). Putative transformants were grown in the greenhouse for molecular analyses. Cre activity in pP2-cre-transformed plants was determined by bombarding T1 embryos with pHK52, which exhibits β-glucuronidase (GUS) activity upon Cre-mediated inversion of the gus coding region. The embryos were stained for GUS activity 48 hr after bombardment.
Molecular Analyses.
Wheat genomic DNA (10 μg), isolated by using the cetyltrimethylammonium bromide method, was subjected to Southern hybridization with 32P-labeled DNA probes. Recombination junctions in the wheat genome were detected by PCR analysis by using primers specific to P1 (5′-CAGCATTGTTCATCGGTAG-3′) and P2 (5′-AGGCTGGCATTATCTACTCG-3′). Intermolecular Cre-lox recombination reactions between PCR-derived junctions and lox-containing DNA fragments were conducted in vitro as described (34). DNA sequencing of PCR fragments was carried out by using the Applied Biosystems Prism automated-sequencing system.
RESULTS AND DISCUSSION
General Strategy.
The transformation vector pVS11 contains a DNA fragment flanked by recombination sites in opposite orientations (Fig. 1A). The lox allele used for the outermost sites is known as lox511, a mutant variant of the wild-type loxP sequence (35). Regardless of the number of copies integrated between the outermost transgenes, and the relative orientation of the outermost pVS11 fragments, recombination between the outermost lox511 sites will resolve multiple units into a single copy. A second set of recombination sites is used for removal of the selectable marker gene, bar, that encodes resistance to the herbicide bialaphos. This second set of loxP sites flanks bar in the same orientation. Because lox511 does not recombine with loxP, two independent recombination reactions are expected to occur. The lox511-to-lox511 recombination will delete internal copies of the transformation DNA to produce a single unit insertion, and the loxP-to-loxP recombination will excise the marker gene. The expected result would be a marker-free, single-copy insertion of the lox511-P1-loxP-P2-lox511 fragment at the integration locus, where P1 and P2 indicate the rice actin and maize ubiquitin gene promoters, respectively (Fig. 1B).
Generation of pVS11 Transformants.
Plasmid pVS11 was delivered into wheat immature embryos through particle bombardment, and cultures were selected on bialaphos. From two transformation experiments, putative transgenic plants were regenerated and their T1 progenies were germinated on selection plates. From the first experiment of ca. 900 immature embryos, 11 putative transgenic lines were regenerated, of which only 2 lines, VS10.1 and VS10.2, yielded resistant T1 seedlings. From the second experiment of more than 1,200 embryos, 25 putative transgenic plants were regenerated, of which 2 lines, I-3 and I-5, yielded resistant T1 seedlings. With each of the four lines, a 3:1 segregation for bialaphos resistance was observed to indicate a single integration locus. The presence of bar in the four lines was confirmed by Southern hybridization.
Generation of cre-Expressing Lines.
The simplest method to introduce Cre recombinase into the system is to cross in a cre-expressing locus. The cre-expressing line was obtained by particle bombardment of ca. 1,200 embryos with pP2-cre and pP2-bar DNA. Both constructs use the maize ubiquitin promoter (P2) to drive expression of cre or bar (Fig. 1A). From the screening of 15 putative transformants, 4 transmitted the resistance trait to progeny. Embryos of these four lines were assayed for Cre activity through bombardment with pHK52, which contains promoter P2 transcribing an antisense gus-coding sequence flanked by oppositely oriented loxP sites. Cre-mediated recombination is expected to invert the gus fragment and permit expression of sense-strand mRNA. The T1 embryos of lines Cre34 and Cre37 stained blue after bombardment with pHK52, with Cre37 as the stronger-expressing line. The presence of bar and cre DNA in Cre37 was confirmed by Southern hybridization. Fortuitously, subsequent analysis of T2 progenies showed that Cre37 contains segregating bar and cre loci, and a Cre37 segregant (Cre37S) was identified that contains cre but lacks the bar locus. Initially, Cre37 and, subsequently, Cre37S were used for crosses with pVS11 transformants.
Marker Excision.
T2 plants of each of the pVS11 lines were crossed with either Cre37 or Cre37S (Table 1). Except for Cre37S, the bar marker is present in all of the parental lines. Genomic DNA of the parents and their F1 progenies were cleaved with EcoRI and hybridized to the bar-coding region (Fig. 1, probe a). The VS10.1, VS10.2, I-3, and I-5 parents all have an internal, 2-kb EcoRI band corresponding to P1-bar (Fig. 2). In representative F1 plants derived from crosses to Cre37 (for VS10.1 and VS10.2) or Cre37S (for I-3 and I-5), this 2-kb P1-bar band is no longer found. This is consistent with Cre-loxP-mediated excision of the bar gene. Cre37 produces an internal, 1.4-kb EcoRI fragment corresponding to P2-bar, and this same band was observed in the F1 progenies derived from crosses with VS10.1 and VS10.2.
Table 1.
Genotype of F2 segregants that harbor pVS11 DNA
| F2 progeny
|
|||||||
|---|---|---|---|---|---|---|---|
| Parents | |||||||
| Female | Male | No. | pVS11 copy | Presence of
|
|||
| donor | donor | F1 line | analyzed | number | cre | P2-bar | amp |
| VS10.1 | Cre37 | A | 4* | 1 | − | − | − |
| 7 | 1 | − | + | + | |||
| 9 | 1 | + | ND | ND | |||
| Cre37 | VS10.1 | B | 13 | 1 | − | + | + |
| 3 | 1 | + | ND | ND | |||
| 4† | M | + | + | + | |||
| VS10.2 | Cre37 | C | 2* | 1 | − | − | − |
| 1 | 1 | − | + | + | |||
| 3 | 1 | + | ND | ND | |||
| 4 | M | + | + | + | |||
| Cre37 | VS10.2 | D | 2 | 1 | − | + | + |
| 6 | 1 | + | ND | ND | |||
| I-3 | Cre37S | E | 1 | 1 | − | − | + |
| 6 | 1 | + | − | + | |||
| 2 | M | + | − | ND | |||
| I-5 | Cre37S | F | 1 | 1 | − | − | + |
| 4 | 1 | + | − | + | |||
F2 progenies are derived from the self-fertilization of a representative F1 plant found to have resolved the multicopy locus of the parent. F2 segregants listed do not include those that segregated away the pVS11 locus. M, multiple copies; ND, not determined.
F3 progeny obtained from outcross to wild type showed the same genotype.
Circular loxP-bar fragment detected.
Figure 2.

Excision of bar gene in F1 progenies. (Upper) EcoRI-cleaved DNA of VS10.1, VS10.2, I-3, I-5, and their representative F1 outcross (to Cre37 or Cre37S) progenies were hybridized to bar (probe a, Fig. 1). (Lower) PCR using primers corresponding to P1 and P2 sequences. Parental DNA shows a 1.8-kb band resulting from the presence of bar between P1 and P2. Representative F1 progenies show a 0.8-kb band from the formation of a new P1-loxP-P2 junction.
The excision of bar should fuse P1 and P2 linked by loxP. Using primers corresponding to P1 and P2, a 0.8-kb fragment corresponding to the new P1-loxP-P2 junction was detected (Fig. 2). In contrast, a band of 1.8 kb representing the P1-loxP-bar-loxP-P2 linkage was amplified from VS10.1, VS10.2, I-3, and I-5, but not from the F1 progenies. A PCR product was not detected from Cre37, Cre37S, or nontransformed plants. The loxP sequence of the 0.8-kb PCR product is functional, because it could recombine with a plasmid substrate in vitro (data not shown). The nucleotide sequence of a representative PCR fragment confirmed that a precise P1-loxP-P2 junction was formed (data not shown).
Reduction of Copy Number and Segregation of the cre Locus.
To identify transgene–host DNA junctions, genomic DNA of parental and progeny lines were probed with a subfragment of the P2 sequence (Fig. 1, probe b). With VS10.1, VS10.2, and I-3, HindIII-cleaved DNA showed intense hybridization to fragments in the 5- to 6-kb range (Fig. 3 A–C). This hybridization is likely to represent multiple copies (in direct and/or inverted orientation) of pVS11, because they are not found in the F1 progenies. The F1 lanes show only bands that could be attributed to the Cre37 or Cre37S genome or to a likely transgene–host DNA junction.
Figure 3.

Reduction of copy number and segregation of cre locus. Genomic DNA of parental lines and their F1 and F2 progenies were cleaved with HindIII (A, B, and C) or EcoRI (D) and hybridized with P2 (probe b, Fig. 1).
Fig. 3 shows representative F2 progenies, derived from self-fertilized F1 plants, which have segregated away the cre locus. In each line, only a single, putative transgene–host DNA junction is found (Fig. 3, arrowheads). For the I-5 line, HindIII-cleaved DNA showed hybridization to high-molecular-weight DNA that migrated at limiting mobility. However, EcoRI-cleaved DNA is well resolved and shows hybridization to two bands of ≈2.5 and ≈3.5 kb (Fig. 3D). If both bands represent transgene–host DNA junctions, this would imply that the two molecules integrated as tandem, inverted copies or in whichever orientation but interspersed by host DNA. In the F1 progeny, a ≈1.6-kb, Cre37S-specific band is found, along with a faint, ≈2.5-kb band and a new, ≈1-kb band. Upon segregation of the cre locus, only this ≈1-kb band remained in the F2 progeny.
Possible Inversion of the Transgene.
Because the resolved, single-copy transgene is flanked by lox511 sites of opposite orientation (Fig. 1B), Cre-mediated recombination between the two sites would invert the transgene. If an inversion were to take place, the P2 hybridization probe would detect a new P2–host DNA junction. This was the case with lines I-3 and I-5. Each F1 plant showed both the original and a new P2–host DNA junction that was not present in its parent. This indicates that the F1 plant was chimeric for the inversion and harbored both transgene orientations. By the next generation, the representative F2 plant chosen for analysis showed only the new P2–host DNA junction. In contrast, transmission of a transgene inversion was not observed for lines VS10.1 and VS10.2. Each F2 plant revealed a P2–host DNA junction fragment of the same size as that detected in the primary transformant.
Target Site Integrity.
F2 or F3 plants that have segregated away the cre locus were analyzed for the expected structure. When cleaved with PstI and probed with P2 DNA (probe b), a 2-kb P2 fragment was observed (Fig. 1B and Fig. 4), consistent with the size in the primary transformants. When cleaved with XhoI and PstI and probed with P1 DNA (Fig. 1B, probe c), a 1.4-kb band representing the full-length P1 fragment was found. In contrast, the parents showed a 2-kb fragment because of the PstI site within the downstream bar gene (Fig. 1A and Fig. 4). The data indicate that both promoters in the progeny plants are intact and are consistent with the intended structure depicted in Fig. 1B.
Figure 4.

Structure of the resolution product. Genomic DNA of F3 or F2 plants derived from each of the parents were cleaved with PstI (Left) and hybridized to P2 (probe b, Fig. 1) or a combination of PstI and XhoI (Right) and hybridized with P1 (probe c, Fig. 1).
The presence of vector DNA was indicated by hybridization to the plasmid-encoded amp gene. Hybridization signals indeed were found in the cre- and bar-deficient F2 segregants derived from I-3 and I-5 (Table 1). Interestingly, of the six F3 segregants derived from VS10.1 and VS10.2 that had segregated away the cre and bar markers (Table 1, F1 lines A and C), none showed hybridization to amp. This could have arisen if all copies of the amp gene were internal to the outermost lox sites and, therefore, were deleted by the Cre-lox reaction.
Chimerism in the Germ Line.
Table 1 summarizes the molecular analysis of F2 segregants derived from six F1 lines that showed resolution of the integration locus. It was surprising to find that 20–40% of the F2 progenies from three lines (Table 1, lines B, C, and E) had maintained multiple copies of the transforming DNA. Thus, although Southern analysis of the F1 plants indicated complete resolution of the integration site, at least when leaf tissues were analyzed, germ-line cells in these three lines must have been chimeric. Furthermore, these F2 progenies show the presence of cre DNA. Whether cre is active in these plants has not been determined.
Rare Instances of an Unexpected Pattern.
The F1 plant derived from the Cre37 ♀ × VS10.1 ♂ cross (Table 1, line B) led to the molecular examination of 20 F2 progenies, of which 4 maintained multiple copies of the transforming DNA. More interestingly, an unexpected band was found when hybridized to bar DNA. The probe detected three EcoRI bands of sizes 1.0, 1.4, and 2.0 kb (Fig. 5, lane 2). The 1.4- and 2.0-kb fragments correspond, respectively, to P2-bar from the Cre37 genome and P1-bar of the unresolved pVS11 integration locus. Incomplete excision is consistent with the detection of multiple copies of pVS11 DNA. When HindIII-cleaved DNA was probed with P2 DNA, the 5-kb fragment characteristic of the unresolved locus was observed. The presence of the 1.0-kb bar band, however, was unexpected. Further analysis suggested that it is a circular loxP-bar molecule (with nos3′; see Fig. 1 legend). Its presence and transmission coincided with the presence of both the unresolved integration locus and P2-cre. Further work is needed to determine why this excision product is maintained whereas most others are lost.
Figure 5.
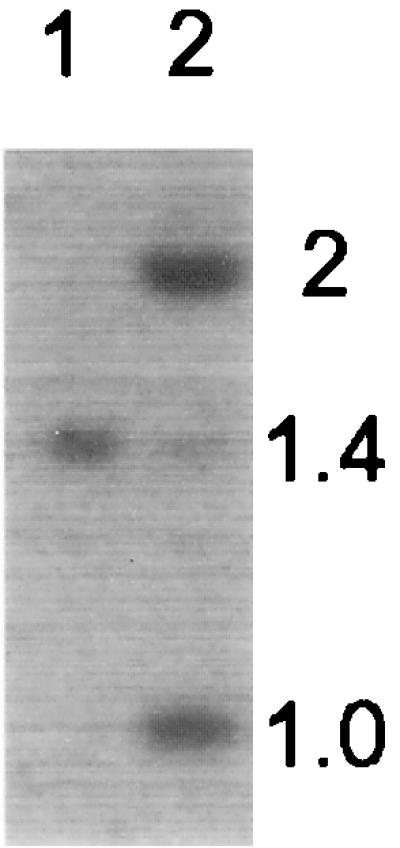
Rare instance of unexpected pattern. Genomic DNA of F2 plants was cleaved with EcoRI and hybridized with bar (probe a, Fig. 1). A majority of F2 plants shows only the 1.4-kb band derived from pP2-bar of the cre genome (lanes 1), but a few individuals show additional bar hybridization bands (lane 2).
Concluding Remarks.
Single-copy transgenic lines are desirable for a variety of reasons. It permits speedier structural and functional characterization, simpler structural documentation, and potentially greater stability in gene structure and expression. The current approach of screening for single-copy insertions by DNA hybridization is capital- and labor-intensive. It is also unpredictable, because success is highly dependent on obtaining a large number of transformants. With the strategy described in this work, the resolution of a complex integration locus through site-specific recombination is expected to produce a unit-copy transgenic plant from virtually 100% of the transformants. This will be of greatest value for plants from which transformants are relatively difficult to obtain. Such is the case with wheat, and with the commercial lines of otherwise transformable plant species, where DNA transformation is often far less efficient than with laboratory cultivars. The direct introduction of DNA into commercial lines would save generations of crosses needed for introgression of the desired trait (39).
Transgene inactivation is affected by several genomic and environmental factors (reviewed in ref. 3). There may be one or several cellular mechanisms triggered by these factors to cause transgene silencing (13, 40). To stabilize gene expression, it may be necessary to control the structure and location of the integration locus in addition to appropriate promoter activity (8, 13). This calls for greater precision in the introduction of DNA molecules. Homologous recombination would be ideal but currently is not a practical option (41). Recombinase-mediated, site-specific integration, however, has shown promise in model plant systems (34, 42). The marker-free resolved sites generated in the present study would be ideal for site-specific insertion of transgenes into the wheat genome.
An important consideration with the current strategy is the frequency of gene copies interspersed with host DNA, as opposed to being in a contiguous, tandem array. Two recent articles have described nontransgenic DNA interspersed among transgene copies in plants (14, 43). However, the origin of the nontransgenic DNA is not known. In mammalian gene-targeting experiments, a process known as “ectopic gene targeting” has been described (44). This is the situation in which the introduced DNA strand invades into the homologous locus and replicates a segment of DNA from that locus. The transgene linked with the copy of host DNA then integrates at a different site. If interspersed DNA were to be found and were derived from elsewhere in the genome, its removal along with the extra transgene copies most likely would be inconsequential. On the other hand, if the nontransgenic DNA were native to the locus, the resolution process would generate a deletion in the chromosome. In light of the latter possibility, for the practical implementation of this strategy, it would be prudent to obtain single-copy transgenic lines from several different progenitor lines.
The pregenome era has been a time in which gene discovery is the limiting factor for crop improvement through genetic engineering. With fewer genes to work with, substantial resources can be devoted to the engineering of fewer traits. For the upcoming postgenome era, however, an exponential increase in the number of genes will be uncovered through genome-sequencing efforts. The functions of a large set of genes will not be known. However, genetic transformation will remain both as a tool for defining gene function and a means for testing the commercial utility of new sequences. With a far greater number of genes to work with, the rate of crop improvement and commercialization of new traits will be much more dependent on the speed and efficiency in testing new DNA sequences in crop genomes. The replacement of conventional screening efforts for single-copy insertions with a resolution-based strategy would be a cost-saving step for the large-scale transformation of new sequences.
Acknowledgments
We thank B. Dowdle-Rizzo and J. Lim for technical assistance, Harold Bockelman for providing plant materials, and H. Koshinsky and H. Albert for pHK52 and pP2-cre, respectively. Funding was provided by Agricultural Research Service CRIS project 5335-21000-009-00D and National Research Initiative Grant NRICGP 94-00952.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Hucl, P, Caswell, K., Demeke, T. & Chibbar, R. N. (1998) Ninth International Wheat Genetics Symposia, August 2–7, 1998, Vol. 3, pp. 189-191.
References
- 1.Assaad F F, Tucker K L, Signer E R. Plant Mol Biol. 1993;22:1067–1085. doi: 10.1007/BF00028978. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson R G, Bieleski L R F, Gleave A P, Janssen B-J, Morris B A M. Plant J. 1998;15:593–604. doi: 10.1046/j.1365-313x.1998.00211.x. [DOI] [PubMed] [Google Scholar]
- 3.Meyer P, Saedler H. Annu Rev Plant Mol Biol. 1996;47:23–48. doi: 10.1146/annurev.arplant.47.1.23. [DOI] [PubMed] [Google Scholar]
- 4.Allen G C, Hall G E J, Childs L C, Weissinger A K, Spiker S, Thompson W F. Plant Cell. 1993;5:603–613. doi: 10.1105/tpc.5.6.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elmayan T, Vaucheret H. Plant J. 1996;9:787–797. [Google Scholar]
- 6.Chareonpornwattana S, Thara K V, Wang L, Datta S K, Pangbangred W, Muthukrishnan S. Theor Appl Genet. 1999;98:371–378. [Google Scholar]
- 7.Meyer P, Linn F, Heidmann I, Meyer A, H, Niedenhof I, Saedler H. Mol Gen Genet. 1992;231:345–352. doi: 10.1007/BF00292701. [DOI] [PubMed] [Google Scholar]
- 8.Finnegan J, McElroy D. Bio/Technology. 1994;12:883–888. [Google Scholar]
- 9.Schuh W, Nelson M R, Bigelow D M, Orum T V, Orth C E, Lynch P T, Eyles P S, Blackhall N W, Jones J, Cocking E C, et al. Plant Sci. 1993;89:69–79. [Google Scholar]
- 10.Srivastava V, Vasil V, Vasil I K. Theor Appl Genet. 1996;92:1031–1037. doi: 10.1007/BF00224045. [DOI] [PubMed] [Google Scholar]
- 11.Matzke A J M, Neuhuber F, Park Y D, Ambros P F, Matzke M A. Mol Gen Genet. 1994;244:219–229. doi: 10.1007/BF00285449. [DOI] [PubMed] [Google Scholar]
- 12.Stam M, deBruin R, Kenter S, van der Hoorn R A L, van Blokland R, Mol J N M, Kooter J M. Plant J. 1997;12:63–82. [Google Scholar]
- 13.Matzke A J M, Matzke M A. Plant Physiol. 1995;107:679–685. doi: 10.1104/pp.107.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohli A, Leech M, Vain P, Laurie D A, Christou P. Proc Natl Acad Sci USA. 1998;95:7203–7208. doi: 10.1073/pnas.95.12.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeBlock M, Debrouwer D, Moens T. Theor Appl Genet. 1997;95:125–131. [Google Scholar]
- 16.Vasil V, Castillo A M, Fromm M E, Vasil I K. Bio/Technology. 1992;10:667–674. [Google Scholar]
- 17.Weeks J T, Anderson O D, Blechl A E. Plant Physiol. 1993;102:1077–1084. doi: 10.1104/pp.102.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker D, Brettschneider R, Lorz H. Plant J. 1994;5:299–307. doi: 10.1046/j.1365-313x.1994.05020299.x. [DOI] [PubMed] [Google Scholar]
- 19.Nehra N S, Chibbar R N, Leung N, Caswell K, Mallard C, Steinhauer L, Baga M, Kartha K K. Plant J. 1994;5:285–297. [Google Scholar]
- 20.Blechl A E, Anderson O D. Nat Bio/Technol. 1996;14:875–879. doi: 10.1038/nbt0796-875. [DOI] [PubMed] [Google Scholar]
- 21.Altpeter F, Vasil V, Srivastava V, Vasil I K. Nat Bio/Technol. 1996;14:1155–1159. doi: 10.1038/nbt0996-1155. [DOI] [PubMed] [Google Scholar]
- 22.Ortiz J P A, Reggiardi M I, Ravizzini R A, Altabe S G, Cervigni G D L, Spitteler M A, Morata M M, Elias F E, Vallegos R H. Plant Cell Rep. 1996;15:877–881. doi: 10.1007/BF00231579. [DOI] [PubMed] [Google Scholar]
- 23.Pedersen C, Zimny J, Becker D, Jahne-Gartner A, Lorz H. Theor Appl Genet. 1997;94:749–757. [Google Scholar]
- 24.Barro F, Rooke L, Bekes F, Gras P, Tatham A S, Fido R, Lazzeri P A, Shewry P R, Barcelo P. Nat Bio/Technol. 1997;15:1295–1299. doi: 10.1038/nbt1197-1295. [DOI] [PubMed] [Google Scholar]
- 25.Barro F, Cannell M E, Lazzeri P A, Barcelo P. Theor Appl Genet. 1998;97:684–695. [Google Scholar]
- 26.Wirtzen B, Brettell R I S, Murray F R, McElroy D, Li Z, Dennis E S. Aust J Plant Physiol. 1998;25:39–44. [Google Scholar]
- 27.Cheng M, Fry J E, Pang S, Zhou H, Hironaka C M, Duncan D R, Conner T R, Wan Y. Plant Physiol. 1997;115:971–980. doi: 10.1104/pp.115.3.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen G, Chilton M-D. Proc Natl Acad Sci USA. 1996;93:14978–14983. doi: 10.1073/pnas.93.25.14978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansen G, Shillito R, Chilton M-D. Proc Natl Acad Sci USA. 1997;94:11726–11730. doi: 10.1073/pnas.94.21.11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dale E C, Ow D W. Proc Natl Acad Sci USA. 1991;88:10558–10562. doi: 10.1073/pnas.88.23.10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoder J I, Goldsbrough A P. Bio/Technology. 1994;12:263–267. [Google Scholar]
- 32.Sauer B. Curr Opin Biotechnol. 1994;5:521–527. doi: 10.1016/0958-1669(94)90068-x. [DOI] [PubMed] [Google Scholar]
- 33.Ow D W. Curr Opin Biotechnol. 1996;7:181–186. [Google Scholar]
- 34.Albert H, Dale E C, Lee E, Ow D W. Plant J. 1995;7:649–659. doi: 10.1046/j.1365-313x.1995.7040649.x. [DOI] [PubMed] [Google Scholar]
- 35.Hoess R H, Wierzbicki A, Abremski K. Nucleic Acids Res. 1986;14:2287–2300. doi: 10.1093/nar/14.5.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McElroy D, Blowers A D, Jenes B, Wu R. Mol Gen Genet. 1991;231:150–160. doi: 10.1007/BF00293832. [DOI] [PubMed] [Google Scholar]
- 37.Christensen A H, Sharrock R A, Quail P H. Plant Mol Biol. 1992;18:675–689. doi: 10.1007/BF00020010. [DOI] [PubMed] [Google Scholar]
- 38.Vasil V, Srivastava V, Castillo A M, Fromm M E, Vasil I K. Bio/Technology. 1993;11:1553–1558. [Google Scholar]
- 39.Christou P. In Vitro Cell Dev Biol. 1993;29P:119–124. [Google Scholar]
- 40.Wasseneger M, Pelissier T. Plant Mol Biol. 1998;37:349–362. doi: 10.1023/a:1005946720438. [DOI] [PubMed] [Google Scholar]
- 41.Puchta H. Trends Plant Sci. 1998;3:77–78. [Google Scholar]
- 42.Vergunst A C, Hooykaas P J J. Plant Mol Biol. 1998;38:393–406. doi: 10.1023/a:1006024500008. [DOI] [PubMed] [Google Scholar]
- 43.Pawlowski W P, Somers D A. Proc Natl Acad Sci USA. 1998;95:12106–12110. doi: 10.1073/pnas.95.21.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dellaire G, Lemieux N, Belmaaza A, Chartrand P. Mol Cell Biol. 1997;17:5571–5580. doi: 10.1128/mcb.17.9.5571. [DOI] [PMC free article] [PubMed] [Google Scholar]