

Abstract
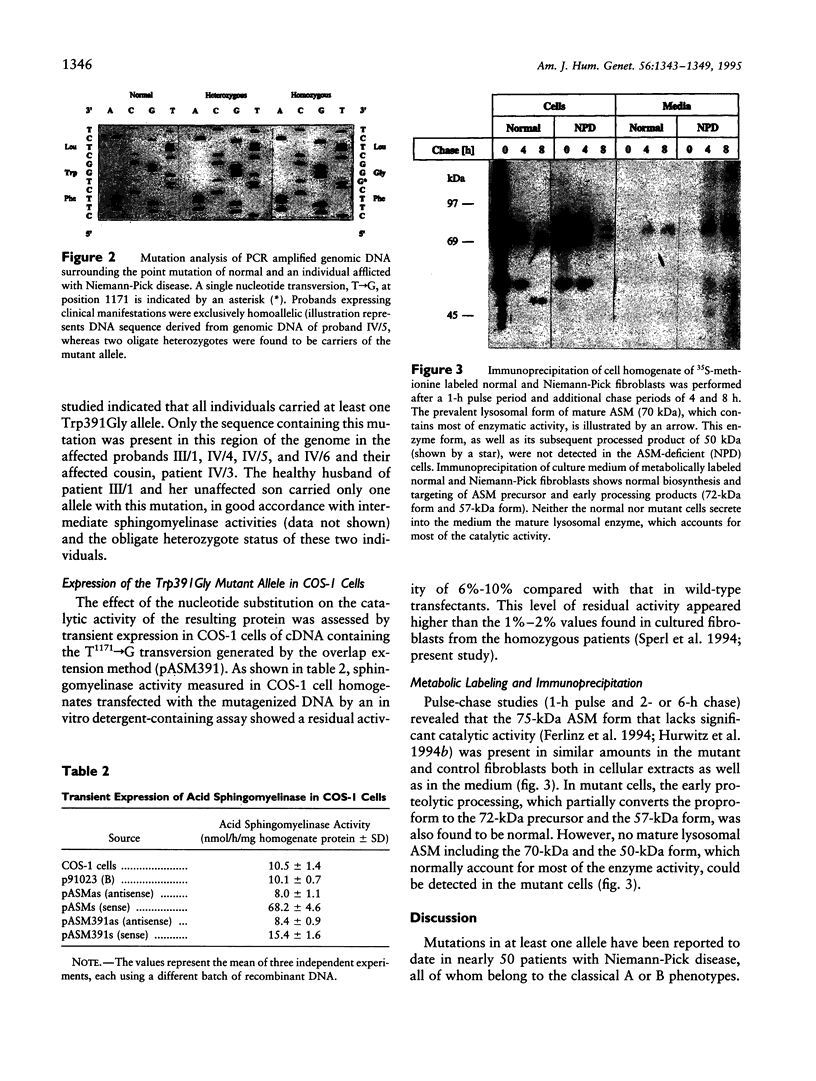
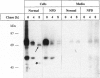
A novel point mutation in the lysosomal acid sphingomyelinase gene has been identified in the recently reported Serbian family with a clinically and biochemically atypical intermediate form of Niemann-Pick disease. The mutation was a T1171-->G transversion resulting in substitution of glycine for normal tryptophan at amino acid residue 391. The coding sequence was otherwise normal. All of the five affected individuals were almost certainly homoallelic, and both of the two obligate heterozygotes studied also carried the same mutation. This mutation is therefore likely to be directly associated with the atypical phenotype of these patients. Expression in COS-1 cells suggested a higher residual activity than that in cultured fibroblasts. A recently developed high-affinity rabbit antihuman sphingomyelinase antibody allowed us to study for the first time the biosynthesis, processing, and targeting of a mutant sphingomyelinase by metabolic labeling of cultured fibroblasts. The mutant enzyme protein was normally synthesized, processed, and routed to the lysosome but was apparently unstable and degraded rapidly once it reached the lysosome. Together with the finding of the relatively high residual activity in COS-1 cells, we interpret our observations to mean that instability and rapid breakdown of the mature mutant enzyme protein, due to the mutation rather than direct inactivation of the catalytic activity, is the primary mechanism for the deficiency of sphingomyelinase activity in these patients. A high prevalence of this mutation in the Serbian population is likely, since the family pedigree indicates that members from four reportedly unrelated families must have contributed the same mutation.
Full text
PDF





Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Albouz S., Hauw J. J., Berwald-Netter Y., Boutry J. M., Bourdon R., Baumann N. Tricyclic antidepressants induce sphingomyelinase deficiency in fibroblast and neuroblastoma cell cultures. Biomedicine. 1981 Dec;35(7-8):218–220. [PubMed] [Google Scholar]
- CROCKER A. C. The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. J Neurochem. 1961 Apr;7:69–80. doi: 10.1111/j.1471-4159.1961.tb13499.x. [DOI] [PubMed] [Google Scholar]
- Ferlinz K., Hurwitz R., Sandhoff K. Molecular basis of acid sphingomyelinase deficiency in a patient with Niemann-Pick disease type A. Biochem Biophys Res Commun. 1991 Sep 30;179(3):1187–1191. doi: 10.1016/0006-291x(91)91697-b. [DOI] [PubMed] [Google Scholar]
- Ferlinz K., Hurwitz R., Vielhaber G., Suzuki K., Sandhoff K. Occurrence of two molecular forms of human acid sphingomyelinase. Biochem J. 1994 Aug 1;301(Pt 3):855–862. doi: 10.1042/bj3010855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989 Apr 15;77(1):51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Hurwitz R., Ferlinz K., Sandhoff K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler. 1994 Jul;375(7):447–450. doi: 10.1515/bchm3.1994.375.7.447. [DOI] [PubMed] [Google Scholar]
- Hurwitz R., Ferlinz K., Vielhaber G., Moczall H., Sandhoff K. Processing of human acid sphingomyelinase in normal and I-cell fibroblasts. J Biol Chem. 1994 Feb 18;269(7):5440–5445. [PubMed] [Google Scholar]
- Kaufman R. J. Identification of the components necessary for adenovirus translational control and their utilization in cDNA expression vectors. Proc Natl Acad Sci U S A. 1985 Feb;82(3):689–693. doi: 10.1073/pnas.82.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levran O., Desnick R. J., Schuchman E. H. Niemann-Pick disease: a frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc Natl Acad Sci U S A. 1991 May 1;88(9):3748–3752. doi: 10.1073/pnas.88.9.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintern L. E., Schuchman E. H., Levran O., Suchi M., Ferlinz K., Reinke H., Sandhoff K., Desnick R. J. Isolation of cDNA clones encoding human acid sphingomyelinase: occurrence of alternatively processed transcripts. EMBO J. 1989 Sep;8(9):2469–2473. doi: 10.1002/j.1460-2075.1989.tb08382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintern L. E., Weitz G., Nehrkorn H., Tager J. M., Schram A. W., Sandhoff K. Acid sphingomyelinase from human urine: purification and characterization. Biochim Biophys Acta. 1987 Dec 14;922(3):323–336. doi: 10.1016/0005-2760(87)90055-5. [DOI] [PubMed] [Google Scholar]
- Rousson R., Bonnet J., Louisot P., Vanier M. T. Presence of immunoreactive material in Niemann-Pick type A placenta using anti-sphingomyelinase rabbit gammaglobulins. Biochim Biophys Acta. 1987 Jun 22;924(3):502–508. doi: 10.1016/0304-4165(87)90166-8. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A. R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977 Dec;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchman E. H., Levran O., Pereira L. V., Desnick R. J. Structural organization and complete nucleotide sequence of the gene encoding human acid sphingomyelinase (SMPD1). Genomics. 1992 Feb;12(2):197–205. doi: 10.1016/0888-7543(92)90366-z. [DOI] [PubMed] [Google Scholar]
- Sperl W., Bart G., Vanier M. T., Christomanou H., Baldissera I., Steichen-Gersdorf E., Paschke E. A family with visceral course of Niemann-Pick disease, macular halo syndrome and low sphingomyelin degradation rate. J Inherit Metab Dis. 1994;17(1):93–103. doi: 10.1007/BF00735404. [DOI] [PubMed] [Google Scholar]
- Takahashi T., Desnick R. J., Takada G., Schuchman E. H. Identification of a missense mutation (S436R) in the acid sphingomyelinase gene from a Japanese patient with type B Niemann-Pick disease. Hum Mutat. 1992;1(1):70–71. doi: 10.1002/humu.1380010111. [DOI] [PubMed] [Google Scholar]
- Takahashi T., Suchi M., Desnick R. J., Takada G., Schuchman E. H. Identification and expression of five mutations in the human acid sphingomyelinase gene causing types A and B Niemann-Pick disease. Molecular evidence for genetic heterogeneity in the neuronopathic and non-neuronopathic forms. J Biol Chem. 1992 Jun 25;267(18):12552–12558. [PubMed] [Google Scholar]
- Vanier M. T., Ferlinz K., Rousson R., Duthel S., Louisot P., Sandhoff K., Suzuki K. Deletion of arginine (608) in acid sphingomyelinase is the prevalent mutation among Niemann-Pick disease type B patients from northern Africa. Hum Genet. 1993 Oct;92(4):325–330. doi: 10.1007/BF01247328. [DOI] [PubMed] [Google Scholar]
- Vanier M. T., Rousson R., Garcia I., Bailloud G., Juge M. C., Revol A., Louisot P. Biochemical studies in Niemann-Pick disease. III. In vitro and in vivo assays of sphingomyelin degradation in cultured skin fibroblasts and amniotic fluid cells for the diagnosis of the various forms of the disease. Clin Genet. 1985 Jan;27(1):20–32. doi: 10.1111/j.1399-0004.1985.tb00180.x. [DOI] [PubMed] [Google Scholar]