

Abstract
Mutations D187N and D187Y in domain 2 of the actin-regulating protein gelsolin cause familial amyloidosis–Finnish type (FAF). We have constructed and expressed a recombinant version of gelsolin domain 2 that is sufficiently stable for kinetic and equilibrium measurements. The wild-type domain and the two amyloidogenic mutants fold via simple two-state kinetics without the accumulation of an intermediate. Unfolding kinetics exhibits significant curvature with increasing urea concentration, indicating that the transition state for unfolding becomes more native-like under increasingly denaturing conditions in accordance with the Hammond postulate. Mutations D187N and D187Y destabilize gelsolin domain 2 by 1.22 and 2.16 kcal⋅mol−1 (1 kcal = 4.18 kJ) respectively. The mutations do not prevent disulfide bond formation despite their direct contiguity with a cysteine residue involved in disulfide linkage. The destabilization conferred on gelsolin domain 2 by the FAF mutations is sufficient to predict that an appreciable fraction is unfolded and, therefore, extremely susceptible to proteolysis at body temperature.
Keywords: protein, stability, denaturation, Hammond, two-state
Defective protein folding is responsible for a wide range of pathological disorders including cancer, emphysema, and cystic fibrosis (1). Experimental evidence increasingly suggests that the amyloid diseases, which include Alzheimer’s and the transmissible spongiform encephalopathies, also result from conformational abnormalities in proteins (2, 3). Amyloidosis is characterized by the extracellular deposition of abnormal protein fibrils that are derived from soluble precursor proteins. Amyloid fibrils appear to have a generic cross-β fibrillar quaternary structure that is independent of the particular disease or protein precursor (4). Moreover, in particular environmental conditions, amyloid fibril formation can be induced in many proteins that are not implicated in amyloid diseases (5). The nature of this unusual arrangement suggests that amyloid formation requires significant structural change in the precursor protein.
Familial amyloidosis–Finnish type (FAF) is an autosomal dominant form of systemic amyloidosis whose phenotype is characterized by corneal lattice dystrophy, cranial neuropathy, skin elasticity problems, and renal complications (6). These symptoms are the result of the accumulation of amyloid fibrils derived from the actin-regulating protein gelsolin (7). Natural gelsolin exists in two forms, which are derived from the same gene by alternative splicing (8): cytoplasmic or intracellular gelsolin is an 84-kDa protein responsible for the assembly and disassembly of actin filaments during events that require cytoskeletal rearrangement, such as fibroblast migration and platelet activation (9). Plasma or secreted gelsolin is 87 kDa in size and contains an additional 25 residues at the N terminus, which may act as a signal sequence for its secretion from the cell. This protein is part of a scavenging system that degrades stray actin filaments in blood plasma and retrieves the constituent actin monomers for intracellular use (10). This function enables gelsolin to reduce the viscosity of pathologically viscous actin-containing materials such as cystic fibrosis sputum and blood clots (11, 12).
Gelsolin knockout studies in mice indicate that the protein is not essential for viability (13), but the severity of the effects vary in different background strains (14). Although the variant gelsolin has defective actin severing activity (15), the pathogenesis of the FAF disease phenotype arises from the accumulation of extracellular amyloid in tissues, rather than the loss of gelsolin function. Plasma gelsolin is the form implicated in FAF (16). Both the cytoplasmic and plasma forms of gelsolin share a 730-aa core structure composed of six homologous domains, G1–6. All six domains have similar fold topology characterized by a central five- to six-strand β-sheet sandwiched between a 3.5–4.5 turn α-helix running parallel to the sheet and a 1–2 turn α-helix perpendicular to the sheet (17).
At the genetic level, FAF is caused by a single nucleotide substitution in the genomic DNA sequence of gelsolin whereby guanine 654 becomes either adenine or thymine (18). This causes the substitution of an aspartate residue at position 187 by asparagine or tyrosine, respectively, which renders gelsolin susceptible to protease digestion at position 173 (Fig. 1). The resulting 68-kDa C-terminal fragment is further digested at residue 243, forming 8.1-kDa peptides whose association gives rise to amyloidogenic deposits (19). The mechanism of amyloid formation remains unclear. Traditionally it has been assumed that mutations in amyloid precursor proteins alter the local surface properties of the native structure favoring different packing interactions for noncovalent polymerization. However, recent evidence suggests that amyloid formation is the result of tertiary conformational change in precursor proteins under partially denaturing conditions, yielding amyloidogenic conformational intermediates with structural features that facilitate fibril formation. Biophysical studies on a number of amyloid precursor proteins have revealed a variety of kinetic and equilibrium amyloidogenic intermediates (20, 21). In this paper, the effects of the disease-causing mutations D187N and D187Y on the folding and stability of gelsolin domain 2 have been examined by using kinetic and equilibrium fluorescence measurements. The amyloidogenic fragment of gelsolin spanning residues 173–243 was recently reported as having negligible residual structure as a monomer in solution (22) and therefore unsuitable for equilibrium and kinetics studies. We find that other constructs of this domain based on crystallographic boundaries are also unfolded or aggregated in solution. Here we have constructed and expressed a protein fragment spanning amino acids 151–266 corresponding to domain 2 of gelsolin with boundaries defined by limited proteolysis. Circular dichroism and one-dimensional NMR spectra (data not shown) indicate that this domain is fully folded as a monomer in solution and is sufficiently stable for equilibrium and kinetic experiments.
Figure 1.

Equilibrium denaturation of gelsolin domain 2. Lines represent the fit of the data to a two-state equation with sloping baselines. (A) Urea titration of wild-type gelsolin 151–266 at 25°C; (B) urea titration of D187N mutant gelsolin 151–266 at 25°C; (C) urea titration of D187Y mutant gelsolin 151–266 at 15°C; and (D) urea titration of wild-type gelsolin 151–266 at 15°C.
MATERIALS AND METHODS
Expression and Purification of Gelsolin 151–266 and Mutants D187N and D187Y.
The amyloid-forming fragment of gelsolin is located within domain 2, whose boundaries have been defined, by limited proteolysis of gelsolin, as histidine-151 to alanine-266 inclusive. This domain contains two tryptophan residues, six trans prolines, and a disulfide bond between cysteine residues 188 and 201. The nucleotide sequence encoding amino acids 151–266 of gelsolin was amplified from the full length human gelsolin gene (23) by PCR with the sense primer GGCCGCGGATCCCACGTGGTACCCAACGAG and the antisense primer CCGCCGGAATTCGGCCGCATCCTCCTTGGC and cloned as a BamHI-EcoRI fragment into the expression vector pRSETA (Invitrogen). Expression vectors containing genes for gelsolin mutants D187N and D187Y were produced from the wild-type gelsolin 151–266 vector by site directed mutagenesis by using the inverse PCR and the following oligonucleotides: TAC TGC TTC ATC CTG GAC C and AAC TGC TTC ATC CTG GAC C are complementary to the noncoding strand of the wild-type gelsolin gene and contain single point mutations (bold type) at their 5′ ends encoding D187Y and D187N, respectively. GCC ATT GTT GAA GCT CTC C is complementary to the coding strand. Constructs were checked by sequencing.
Gelsolin 151–266 and D187N and D187Y mutants were expressed in BL21(C41) Escherichia coli cells (24) transformed with the appropriate vector. Cultures were grown in LB-broth with ampicillin (150 μg/ml) at 37°C and induced with isopropyl-d-thiogalactoside at A600 = 0.4. Cells were harvested by centrifugation. The cell pellets were resuspended in Pi buffer (20 mM phosphate, pH 7.2 with NaCl to 0.2 M calculated ionic strength, 100 ml per liter of LB culture). The cells were lysed by sonication, and cell debris was pelleted by centrifugation. The supernatant was purified by using nickel affinity chromatography, and thrombin was used to cleave the histidine tag. A final gel-filtration step removed residual impurities and the soluble aggregated gelsolin fraction that eluted within the void volume of the column. Fractions containing monomeric gelsolin were dialyzed against 50 mM Tris⋅HCl, pH 7.2/150 mM NaCl and stored at 4°C. Protein concentrations were determined by measuring the absorbance at 280 nm by using an extinction coefficient of 12,900 M−1 cm−1 estimated by the method of Gill and von Hippel (25). Protein was checked by using mass spectrometry. Ellman’s assay for free thiols was performed (26).
Equilibrium Fluorescence Measurements.
Stock solutions of urea (8 or 9 M) in buffer (50 mM Tris⋅HCl, pH 7.2/150 mM NaCl) were made up gravimetrically. A range of 67 concentrations of denaturant (800 μl of each concentration in a 1.5-ml Eppendorf tube) was produced by dilution of stock solution with buffer by using a Hamilton Microlab. An aliquot (100 μl) of filtered (0.2 μm Sartorius Minisart filter) protein in the same buffer was added to each sample of denaturant, giving a final protein concentration of 2 μM. The samples were preequilibrated at the required temperature for at least 2 hr. Fluorescence of each sample was then measured by using an Aminco Bowman Series 2 luminescence spectrometer with excitation at 280 nm, emission at 356 nm, and band passes of 4 nm. A scan of the fluorescence was taken from 300–400 nm with samples thermostated at 25 or 15°C. Data sets were fitted to standard two-state equations with sloping baselines (27).
Stopped-Flow Fluorescence Measurements.
The kinetics of gelsolin 151–266 and mutants D187N and D187Y refolding and unfolding was monitored by using an Applied Photophysics (Surrey, U.K.) SF.17MF stopped-flow apparatus thermostated at 25 or 15°C. All experiments were performed in 50 mM Tris⋅HCl, pH 7.2, with 150 mM NaCl. For unfolding, 33 μM protein in buffer was mixed at a ratio of 1:10 with buffer containing a range of urea concentrations. For refolding, 33 μM protein denatured with 5.5 M urea in buffer was mixed at a ratio of 1:10 with buffer containing a range of urea concentrations. In addition, 6 μM protein denatured in 32 mM HCl with 150 mM NaCl was mixed at a ratio of 1:1 with 100 mM Tris⋅HCl, pH 8.0, with 150 mM NaCl and either 0 or 0.5 M urea, yielding a final pH of 7.2. Fluorescence intensity on mixing was monitored with excitation at 280 nm and emission detection above 320 nm by using a glass cut-off filter. Data were acquired for at least 10 half-lives of the slowest phase and fitted to exponential equations by using the nonlinear regression analysis program kaleidagraph [Version 2.1, Synergy Software (Reading, PA)].
RESULTS
Equilibrium Results.
Urea titration of wild-type gelsolin 151–266 monitored by fluorescence shows cooperative unfolding and refolding transitions between 1 and 3.5 M urea at 25°C (Fig. 1A). The entire data set from the fluorescence monitored denaturation experiments was fitted to a two-state equation with sloping baselines (27). The values of [urea]50%, the urea concentration at which the native and denatured states are populated equally, and m, the dependence of the free energy of unfolding on denaturant concentration, are listed in Table 1. The values for ΔGD–NH2O are obtained from the product of [urea]50% and m. Unfolding by chemical denaturation is reversible (data not shown).
Table 1.
Values obtained from equilibrium fluorescence titration of gelsolin domain 2
| Protein | T, °C | [urea]50%, M | m, kcal·mol−1 | ΔGD–NH2O, kcal·mol−1 |
|---|---|---|---|---|
| Wild type | 25 | 2.28 ± 0.04 | 1.72 ± 0.12 | 3.94 ± 0.19 |
| Wild type | 15 | 2.82 ± 0.02 | 1.67 ± 0.03 | 4.71 ± 0.06 |
| D187N | 25 | 1.59 ± 0.11 | 1.92 ± 0.32 | 3.06 ± 0.43 |
| D187Y | 15 | 1.60 ± 0.14 | 1.75 ± 0.35 | 2.81 ± 0.48 |
At 25°C, urea titration of D187N monitored by fluorescence shows a cooperative unfolding transition between 0.5 and 2.5 M urea. The fitted values (Table 1) obtained from this plot (Fig. 1B) indicate that gelsolin 151–266 incurs a loss of free energy in the presence of mutation D187N with ΔΔG = 1.22 ± 0.17 kcal⋅mol−1 (1 kcal = 4.18 kJ) [calculated by using an average equilibrium m value multiplied by Δ[urea]50% (28)]. Urea titration of D187Y at 25°C produced only a partial denaturation curve, demonstrating that the protein was not fully folded in water at this temperature (data not shown). However, at 15°C, a cooperative unfolding transition between 0.5 and 2.5 M urea was observed for D187Y (Fig. 1C), showing a destabilization of the domain by 2.16 ± 0.20 kcal⋅mol−1 relative to wild type at 15°C (Fig. 1D).
Stopped-Flow Kinetics.
The change in tryptophan fluorescence between the folded and denatured states was used to monitor folding and unfolding rates after rapid mixing of protein and varying concentrations of denaturant. All of the unfolding curves fit well to single exponential equations. Refolding kinetics displays a fast phase that accounts for 70% of the amplitude of the fluorescence change on refolding, and only this phase is considered here. The remaining 30% of the amplitude comprises three slower phases thought to be the result of cis-trans proline isomerization about peptidyl-prolyl bonds; gelsolin 151–266 contains six trans prolines in the native state. Refolding data were fitted to a quadruple exponential equation with a nonzero end point and a drift term by first fitting the tail of the process to a double exponential equation and then fitting the whole curve to a quadruple exponential in which the last two phases had fixed rate constants and amplitudes (29).
Unfolding Kinetics.
The plots of lnkU vs. [urea] for unfolding of gelsolin 151–266 and mutants D187N and D187Y exhibit significant curvature (Fig. 3). Such nonlinearity has been observed for other proteins and may result from small movements in the position of the transition state with increasing denaturant concentration (30, 31). To account for this nonlinearity, the data were fitted to a second-order polynomial equation:
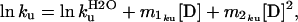 |
1 |
where m1kuand m2ku are the coefficients for the first- and second-order [urea] terms respectively. The slope of the plot at a given urea concentration is given by:
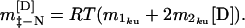 |
2 |
Figure 3.

Effects of disulfide bond formation on gelsolin domain 2. Lines represent the fit of the data to a two-state equation with sloping baselines. Urea titration of wild-type gelsolin 151–266 at 25°C in the presence (unfilled circles) and absence (filled circles) of 1 mM DTT.
Refolding Kinetics.
The plots of lnkf vs. [urea] for the refolding of gelsolin 151–266 and the D187N and D187Y mutants exhibit slight deviations from linearity (Fig. 2 B and C). Hence the data were fitted to a second-order polynomial equation:
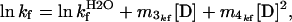 |
3 |
where m3kf and m4kf are the coefficients for the first and second-order [D] terms respectively. The slope of the plot at a particular urea concentration is given by
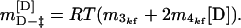 |
4 |
The complete kinetics of folding and unfolding can be fitted to Eq. 5, which is derived from Eqs. 1 and 3.
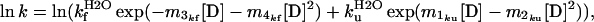 |
5 |
where k is the observed rate of unfolding or refolding at a particular denaturant concentration. The kinetic data for urea unfolding and refolding of gelsolin 151–266 and mutants D187N and D187Y can be fitted to this two-state model to yield the values for kfH2O, kuH2O, m1ku, m2ku, m3kf and m4kf (Table 2).
Figure 2.

Kinetics of folding and unfolding of gelsolin domain 2. The curve indicates the best fit of the data to Eq. 5, where y = ln(kobs) and x is the denaturant concentration. (A) Chevron plot for wild-type gelsolin 151–266 at 25°C; (B) chevron plot for D187N mutant gelsolin 151–266 at 25°C; and (C) chevron plot for D187Y mutant gelsolin at 15°C.
Table 2.
Fitted values obtained from stopped-flow fluorescence studies on gelsolin domain 2
| Protein | T, °C | kuH2O | m1ku | m2ku | kfH2O | m3kf | m4kf |
|---|---|---|---|---|---|---|---|
| Wild type | 25 | 0.058 ± 0.01 | 1.19 ± 0.09 | −0.064 ± 0.008 | 28.6 ± 1.19 | 1.03 ± 0.09 | 0.24 ± 0.05 |
| D187N | 25 | 0.420 ± 0.06 | 1.04 ± 0.06 | −0.045 ± 0.006 | 25.2 ± 0.68 | 1.06 ± 0.10 | 0.46 ± 0.07 |
| D187Y | 15 | 0.151 ± 0.03 | 1.29 ± 0.09 | −0.065 ± 0.010 | 12.5 ± 0.50 | 1.02 ± 0.12 | 0.33 ± 0.07 |
Comparison of Equilibrium and Kinetic Data.
The free energy of unfolding at zero denaturant concentration can be estimated from the above kinetic data by using Eq. 6:
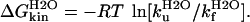 |
6 |
Similarly, an m value can be predicted from kinetic data:
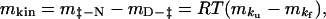 |
7 |
where m‡-N = δΔG‡-N/δ[urea] and mD-‡ = δΔGD-‡/δ[urea] over the ranges of [urea] used in the experiment (29). Values of ΔGkinH2O and mkin for gelsolin 151–266 and mutants D187N and D187Y are shown in Table 3. All values of ΔGkinH2O and mkin obtained from kinetic data correspond within error to the equivalent values derived from equilibrium experiments.
Table 3.
Values obtained from stopped-flow fluorescence studies on gelsolin domain 2
| Protein | T, °C | [urea]50%, M | m*, kcal·mol−1 M−1 | ΔGkinH2O†, kcal·mol−1 |
|---|---|---|---|---|
| Wild type | 25 | 2.65 | 1.78 ± 0.10 | 3.67 ± 0.16 |
| D187N | 25 | 1.85 | 2.02 ± 0.09 | 2.43 ± 0.19 |
| D187Y | 15 | 1.82 | 1.80 ± 0.01 | 2.62 ± 0.19 |
Calculated from mkin = m‡-N − mD-‡ = RT(mku − mkf).
Calculated from ΔGkinH2O = −RT ln [kuH2O/kfH2O].
Studies on the Disulfide Bond.
Gelsolin domain 2 contains a disulfide bond between cysteine residues 188 and 201. The proximity of cysteine-188 to aspartate-187 has prompted the suggestion that the FAF disease mutations, D187N and D187Y, inhibit formation of the disulfide bond leaving gelsolin vulnerable to proteolytic attack (32). Ellman’s assay for thiols was used to demonstrate that the disulfide bond is fully formed in the wild-type and D187N and D187Y mutant versions of gelsolin domain 2 used in this paper. The mutations do not prevent disulfide bond formation.
Equilibrium urea denaturation experiments were carried out on wild-type (Fig. 3) and D187Y mutant protein in the presence of the reducing agent DTT to investigate the effects of removing the disulfide bond on folding and stability. Interestingly, although [urea]50% was reduced from 2.28 ± 0.04 M in wild-type gelsolin 151–266 to 1.64 ± 0.04 M when the disulfide was removed by using DTT, the m value was seen to increase from 1.72 ± 0.12 kcal⋅mol−1 M−1 to 2.80 ± 0.02 kcal⋅mol−1 M−1. These fitted parameters yield a value of ΔGD–NH2O equal to 4.59 ± 0.03 kcal⋅mol−1, which is higher than the value of 3.67 ± 0.16 kcal⋅mol−1 obtained when the disulfide bond is present. Equilibrium denaturation of mutant D187Y at 15°C in the presence of DTT produced a data set with no folded baseline. Therefore no fitted parameters could be determined. However, the visible difference between the denaturation curves obtained for mutant D187Y in the presence and absence of DTT lend further support to the fact that the disulfide bond is formed correctly despite the mutation in the protein.
DISCUSSION
Gelsolin Domain 2 Folds via a Two-State Mechanism.
The FAF disease-causing mutations, D187N and D187Y, found in gelsolin, effect a sequence of physiological events that culminate in extracellular amyloid deposition. It is clear that mutant gelsolin is aberrantly cleaved at two positions, yielding the amyloidogenic fragment comprising residues 173–243. However, there are still many gaps in the current knowledge of the disease mechanism. A biophysical analysis of the folding behavior of gelsolin domain 2 and mutants D187N and D187Y is described here. All values for ΔGD–NH2O and m obtained from kinetic data are equivalent within error to the corresponding values derived from equilibrium experiments (Table 1), indicating that each of the three proteins folds via a two-state mechanism (33). This suggests that within the limitations of these experiments, the FAF disease-causing mutations do not affect the folding pathway of gelsolin domain 2. There is no evidence for the accumulation of on- or off-pathway conformational intermediates in either the wild-type or mutant proteins, although these experiments do not rule out the possibility of transient high-energy intermediates.
Is the Proteolytic Susceptibility of Gelsolin Mutants D187N and D187Y the Result of Instability?
The mutations D187N and D187Y destabilize the protein by 1.22 and 2.16 kcal⋅mol−1, respectively. Values for the equilibrium constant Keq can be estimated from ΔGD–NH2O = RTlnKeq and Keq = N/D at equilibrium. At 25°C, Keq for wild-type and mutant D187N is equal to 777 and 176, respectively. At 15°C, mutant D187Y has Keq = 136, compared with 3,768 for wild type, which translates to a 25-fold increase in population of the denatured state in 0 M denaturant. At 37°C, the temperature of the human body, this effect is expected to be further pronounced, rendering a significant fraction of the gelsolin of a FAF patient at least partially unfolded under physiological conditions. This may allow proteolytic enzymes access to sites that are protected in the folded protein. Furthermore, cleavage and export of unfolded gelsolin may result in additional unfolding of native gelsolin to maintain the equilibrium between the native and denatured states.
Conformational Instability.
As domain 2 folds by a simple two-state cooperative transition, the destabilization of the protein by mutations at position 187 causes a global destabilization of the whole domain, which can lead to proteolysis. In addition, local instability may be because the mutations disrupt the network of interactions around the carboxylate of aspartate-187. This may well render the site susceptible to proteolysis without global unfolding.
FAF Disease-Causing Mutations Do Not Prevent Disulfide Bond Formation.
Because the site of the FAF disease-causing mutations, aspartate-187, is directly adjacent to one of the cysteine residues involved in disulfide bond formation, it has been suggested that the mutations disrupt the formation of the disulfide linkage in gelsolin domain 2, leaving the protein exposed to abnormal proteolytic attack. Paunio et al. (32) found that disrupting the disulfide bond in gelsolin domain 2 by site-directed mutagenesis led to aberrant intracellular proteolysis comparable to that observed in FAF, which appears to support this theory. However, the results presented here indicate that it is possible for the disulfide bond to form correctly despite the mutation. The presence of a disulfide bond limits the conformational entropy of the denatured state. Therefore the concentration of urea required to denature gelsolin domain 2 is reduced on reduction of the disulfide bond by using DTT. However, there is a larger difference in solvent-accessible surface area between the native and denatured states when the disulfide is absent, which is experimentally observed as a higher m value. This is not unexpected, because the disulfide forms between the C and D strands of the central β-sheet (17). The fact that the disulfide bond can form correctly despite the mutation suggests that the proteolytic susceptibility of the mutant protein is not caused by the absence of the disulfide.
Comparison with Villin 14T.
The folding kinetics of villin 14T, a domain that shares significant sequence homology and fold topology with gelsolin domain 2, was recently published (34). Despite its similarity to gelsolin domain 2, villin 14T is significantly more stable, having ΔGD–NH2O = 9.7 kcal⋅mol−1 at 25°C, compared with 3.9 kcal⋅mol−1 for gelsolin domain 2 under the same experimental conditions. In addition, villin 14T exhibits an off-pathway folding intermediate in urea at 25°C but appears to fold via a two-state mechanism at 37°C.
Some Other Amyloid Precursor Proteins Form Conformational Intermediates.
Transthyretin, the protein responsible for senile systemic amyloidosis and the familial amyloid polyneuropathies, unfolds via an intermediate with a defined β-sheet-rich tertiary structure in acidic conditions (35). Moreover, mutant lysozyme variants form molten globule-like intermediates on their thermal denaturation pathway (36). In contrast, the Aβ peptide, which is the amyloidogenic determinant of Alzheimer’s disease, appears to gain structure on the path to amyloidosis (37). Like gelsolin’s amyloidogenic fragment, this peptide is formed from the proteolytic cleavage of a larger parent protein. Ratnaswamy et al. (22) have recently demonstrated that the amyloidogenic fragment of gelsolin, residues 173–243, is largely unstructured when isolated in vitro and that proteolysis is necessary but not sufficient for amyloid formation. An environmental change is further required for amyloid construction from the proteolytic fragment. These observations complement the suggestion that it is the denatured fraction of mutant gelsolin that is susceptible to proteolysis. It appears that, although the amyloid fibrils from different diseases have a common structure, the events leading to their formation are varied, at least in the early stages.
ABBREVIATION
- FAF
familial amyloidosis–Finnish type
References
- 1.Perrett S. Chem Ind (London) 1998;10:389–393. [Google Scholar]
- 2.Kelly J W. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 3.Kelly J. Structure (London) 1997;5:595–600. doi: 10.1016/s0969-2126(97)00215-3. [DOI] [PubMed] [Google Scholar]
- 4.Blake C, Serpell L. Structure (London) 1996;4:989–998. doi: 10.1016/s0969-2126(96)00104-9. [DOI] [PubMed] [Google Scholar]
- 5.Chiti F, Webster P, Taddei N, Clark A, Stefani M, Ramponi G, Dobson C M. Proc Natl Acad Sci USA. 1999;96:3590–3594. doi: 10.1073/pnas.96.7.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meretoja J. Ann Clin Res. 1969;1:314–324. [PubMed] [Google Scholar]
- 7.Haltia M, Prelli F, Ghiso J, Kiuru S, Somer H, Palo J, Frangione B. Biochem Biophys Res Commun. 1990;167:927–932. doi: 10.1016/0006-291x(90)90612-q. [DOI] [PubMed] [Google Scholar]
- 8.Kwiatkowski D J, Stossel T P, Orkin S H, Mole J E, Colten H R, Yin H L. Nature (London) 1986;323:455–458. doi: 10.1038/323455a0. [DOI] [PubMed] [Google Scholar]
- 9.Stossel T P. Sci Am. 1994;271:54–63. doi: 10.1038/scientificamerican0994-54. [DOI] [PubMed] [Google Scholar]
- 10.Lee W, Galbraith R. N Engl J Med. 1992;326:1335–1341. doi: 10.1056/NEJM199205143262006. [DOI] [PubMed] [Google Scholar]
- 11.Vasconcellos C A, Allen P G, Wohl M E, Drazen J M, Janmey P A, Stossel T P. Science. 1994;263:969–971. doi: 10.1126/science.8310295. [DOI] [PubMed] [Google Scholar]
- 12.Davoodian K, Ritchings B, Ramphal R, Bubb M. Biochemistry. 1997;36:9637–9641. doi: 10.1021/bi9711487. [DOI] [PubMed] [Google Scholar]
- 13.Witke W, Kwiatkowski D J. Cell. 1995;81:41–51. doi: 10.1016/0092-8674(95)90369-0. [DOI] [PubMed] [Google Scholar]
- 14.Kwiatkowski D J. Curr Opin Cell Biol. 1999;11:103–108. doi: 10.1016/s0955-0674(99)80012-x. [DOI] [PubMed] [Google Scholar]
- 15.Weeds A G, Gooch J, McLaughlin P, Maury C P J. FEBS Lett. 1993;335:119–123. doi: 10.1016/0014-5793(93)80452-z. [DOI] [PubMed] [Google Scholar]
- 16.Kangas H, Paunio T, Kalkkinen N, Jalanko A, Peltonen L. Hum Mol Genet. 1996;5:1237–1243. doi: 10.1093/hmg/5.9.1237. [DOI] [PubMed] [Google Scholar]
- 17.Burtnick L D, Koepf E K, Grimes J, Jones E Y, Stuart D I, McLaughlin P J, Robinson R C. Cell. 1997;90:661–670. doi: 10.1016/s0092-8674(00)80527-9. [DOI] [PubMed] [Google Scholar]
- 18.de la Chapelle A, Tolvanen R, Boysen G, Santavy J, Bleeker-Wagemakers L, Maury C P, Kere J. Nat Genet. 1992;2:157–160. doi: 10.1038/ng1092-157. [DOI] [PubMed] [Google Scholar]
- 19.Maury C, Sletten K, Totty N, Kangas H, Liljestrom M. Lab Invest. 1997;77:299–304. [PubMed] [Google Scholar]
- 20.Kelly J W. Curr Opin Struct Biol. 1998;8:101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 21.Wetzel R. Cell. 1996;86:699–702. doi: 10.1016/s0092-8674(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 22.Ratnaswamy G, Koepf E, Bekele H, Yin H, Kelly J W. Chem Biol. 1999;6:293–304. doi: 10.1016/S1074-5521(99)80075-1. [DOI] [PubMed] [Google Scholar]
- 23.Way M, Gooch J, Pope B, Weeds A G. J Cell Biol. 1989;109:593–605. doi: 10.1083/jcb.109.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miroux B, Walker J E. J Mol Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 25.Gill S C, von Hippel P H. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 26.Ellman G L. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 27.Clarke J, Fersht A R. Biochemistry. 1993;32:4322–4329. doi: 10.1021/bi00067a022. [DOI] [PubMed] [Google Scholar]
- 28.Serrano L, Kellis J T, Jr, Cann P, Matouschek A, Fersht A R. J Mol Biol. 1992;224:783–804. doi: 10.1016/0022-2836(92)90562-x. [DOI] [PubMed] [Google Scholar]
- 29.Fersht A R. Structure and Mechanism in Protein Science. San Francisco: Freeman; 1998. [Google Scholar]
- 30.Matouschek A, Fersht A R. Proc Natl Acad Sci USA. 1993;90:7814–7818. doi: 10.1073/pnas.90.16.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matouschek A, Otzen D E, Itzhaki L S, Jackson S E, Fersht A R. Biochemistry. 1995;34:13656–13662. doi: 10.1021/bi00041a047. [DOI] [PubMed] [Google Scholar]
- 32.Paunio T, Kangas H, Heinonen O, Buc-Caron M H, Robert J J, Kaasinen S, Julkunen I, Mallet J, Peltonen L. J Biol Chem. 1998;273:16319–16324. doi: 10.1074/jbc.273.26.16319. [DOI] [PubMed] [Google Scholar]
- 33.Jackson S E, Fersht A R. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 34.Choe S E, Matsudaira P T, Osterhout J, Wagner G, Shakhnovich E I. Biochemistry. 1998;37:14508–14518. doi: 10.1021/bi980889k. [DOI] [PubMed] [Google Scholar]
- 35.Lai Z, Colon W, Kelly J W. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 36.Booth D R, Sunde M, Bellotti V, Robinson C V, Hutchinson W L, Fraser P E, Hawkins P N, Dobson C M, Radford S E, Blake C C F, et al. Nature (London) 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 37.Lee J P, Stimson E R, Ghilardi J R, Mantyh P W, Lu Y A, Felix A M, Llanos W, Behbin A, Cummings M, Van Criekinge M, et al. Biochemistry. 1995;34:5191–5200. doi: 10.1021/bi00015a033. [DOI] [PubMed] [Google Scholar]