

Abstract
The recombinant bovine factor B, obtained by a newly developed bacterial expression system, was found to exhibit features characteristic of a reversible self associating system. Using size-sieving chromatography, distribution of the factor B species ranged from a monomer to a trimer, but not oligomers of higher molecular weights. At high protein concentrations, factor B migrated as a single band in a native gel. Cross-linking with the amino-reactive cross-linking reagent bis (sulfosuccinimidyl) suberate (BS), at a low cross-linker to protein ratio yielded cross-linked products identified as factor B dimer and trimer. The cross-linking pattern was shown to be a function of the protein and cross-linker concentrations. The range of sedimentation coefficients in a sedimentation velocity experiment suggested that the largest particle present in the distribution was more than twice as large as the smallest. The data obtained under multiple conditions in the sedimentation equilibrium experiments are best fit to a model describing a reversible self association of a monomer-trimer of factor B species, with a dissociation constant Kd1,3 = 2.48×10-10 M2.
INTRODUCTION
Factor B is a peripheral membrane protein, removal of which from the mitochondrial inner membrane by a treatment of well-coupled bovine heart mitochondria using ultrasound at pH ∼8.8 in the presence of EDTA renders the ammonia, EDTA-treated submitochondrial particles (AE-SMP)3 incapable of catalyzing the partial reactions of oxidative phosphorylation, which require the electrochemical proton potential Δμ H+ as an intermediary [1-5]. The inability of AE-SMP to form and maintain a Δμ H+ is due to a proton leak, which uses, at least in part, the proton-translocating pathway of the ATP synthase complex membrane sector F0, because the proton leak has been shown to be blocked by substoichiometric amounts of the F0 inhibitor oligomycin [6], albeit less potently than by factor B [3].
We recently cloned and expressed both the human [3] and bovine [5] factor B polypeptides, and demonstrated that reconstitution of the bovine AE-SMP, which are essentially depleted of endogenous factor B, with either recombinant polypeptide, restored ΔμH+g-dependent reactions catalyzed by the vesicles to the levels observed in coupled SMP [3-5]. The expression systems used for overproducing the recombinant factor B in Escherichia coli utilized constructs in which the bacterial polypeptides thioredoxin or NusA were fused to the NH2-termini of human or bovine factor B, respectively, separated by a linker [3-5]. The expression of the thioredoxin-human factor B or NusA-bovine factor B fusion polypeptides were regulated by the arabinose or T7 promoters, respectively. Although a significant portion of the factor B fusion polypeptides was found in a soluble fraction of the E. coli lysate with each of these two systems, the fidelity of overproduction varied significantly between them. The final yields of the recombinant human and bovine factor B polypeptides, following fusion moiety removal, were roughly 1 mg and 12-15 mg of factor B per liter of bacterial culture, respectively. Because the human and bovine polypeptides exhibit 90% sequence identity [5], a difference in the yields of the recombinant polypeptides overexpression from the pBAD/Thio-TOPO and pET-43.1a vectors is likely related to a difference in the robustness of the two expression systems.
When purifying the recombinant bovine factor B, we noted a tendency of the protein to oligomerize, following removal of the NusA moiety. We used a combination of chromatographic, electrophoretic and chemical cross-linking approaches, as well as a thorough analytical ultracentrifugation analysis, to characterize the oligomerization properties of the recombinant bovine factor B in solution. Herein we report that, using size-sieving chromatography, a predominant portion of the recombinant polypeptide eluted with a relative molecular weight of ∼56 kDa, and a minor portion of the polypeptide eluted with a relative molecular weight of ∼34 kDa. Using a calculated molecular mass of the recombinant bovine factor B of 20,412 Da, these polypeptides could represent trimer and dimer species of factor B, respectively. Chemical cross-linking with the amino-group specific water-soluble cross-linking reagent bis (sulfosuccinimyidyl) suberate (BS), with a spacer arm of 11.4 Å, provided additional evidence for the existence of factor B oligomers in solution, and demonstrated that the factor B cross-linking pattern was a function of the protein and the cross-linker concentrations. Subsequent sedimentation velocity experiments yielded bovine factor B sedimentation coefficients that ranged between 1.7 s and 3.85 s, suggesting that the largest particle is more than twice as large as the smallest particle in the distribution, supporting a monomer-trimer model. Finally, the data obtained under multiple conditions in the sedimentation equilibrium experiments are best fit to a model describing a reversible self association of a monomer-trimer of factor B, with a dissociation constant Kd1,3 = 2.48 × 10-10M2.
MATERIALS AND METHODS
Materials
NAD, NADH, ATP, oligomycin, 2,4-dithiothreitol, and IPTG were obtained from EMD Biosciences-Calbiochem; Sephacryl S-200 HR and DEAE-Sepharose FF were purchased from GE Healthcare; BS, EDC, sulfo-NHS, sulfo-DST were from Pierce; 30% acrylamide/bis solution, 37.5:1, low molecular weight standards, and Macro-Prep High Q anion exchange resin were from Bio-Rad Laboratories; 7% Tris-Acetate NuPAGE gels were purchased from Invitrogen. All other chemicals were reagent grade. The AE-SMP were prepared from heavy beef heart mitochondria, as described previously [3].
Assays
Standard molecular biology techniques, including cloning, restriction enzyme analysis, plasmid preparation, and agarose gel electrophoresis were performed as described previously [7].
The ATP-driven reverse electron transfer from succinate to NAD was assayed at 38°C essentially according to [8], except that after 1-2 min of incubation of AE-SMP with factor B, succinate, ATP, MgCl2, KCN, and dithiothreitol, the reaction was started by the addition of NAD. The reaction was monitored at 340 nm in a spectrophotometer, and the rate of NAD reduction was calculated using an extinction coefficient of 6.22 mM-1 cm-1.
The cross-linking of factor B with BS was done as follows. Briefly, the factor B storage buffer was exchanged for a pH 7.8 buffer containing 50 mM HEPES and 0.1 M NaCl by centrifugation through the macro spin P-6 column (Harvard Apparatus, Holliston, MA) equilibrated with the same buffer. The protein concentration was determined by a BCA reagent (Pierce) or by absorption at 280 nm, using the molar extinction coefficient ε280 of 31160 M-1 cm-1 for the recombinant bovine factor B as determined from sequence by the method of McGill and von Hippel [9]. Freshly prepared stock solution of BS in the above buffer was added to the factor B solution at concentrations indicated in the figure legends and the reactions proceeded for 5 and 10 min at room temperature. The reactions were terminated with 100 mM ammonium acetate, followed by the addition 5 min later of the SDS-PAGE sample buffer. The cross-linking of factor B with BS was analyzed by SDS-PAGE [10] under reducing conditions using gels of various polyacrylamide concentrations as indicated in the figure legends.
Expression and purification of recombinant bovine factor B
The recombinant bovine factor B was expressed and purified as described earlier [5], with the following modifications. 1. After the bacterial culture reached an OD of 0.5-0.6 at 600 nm at 37°C, the temperature of the shaker was shifted to 25°C, flasks with the bacterial culture were allowed to equilibrate at the indicated temperature, and protein expression was induced with 1 mM IPTG for 18 hrs. 2. Following the size-sieving chromatography on Sephacryl S-200 HR and the ion-exchange chromatography on Macro-Prep High Q anion exchange resin, the NusA-bovine factor B fusion polypeptide was salted out with 50% saturated ammonium sulfate (29.1 g per 100 ml protein solution), stirred on ice slurry for 15 min, then pelleted by centrifugation at 14,000 rpm for 15 min at 4°C. 3. At the final step of the recombinant factor B purification, a 0-0.3 M NaCl gradient was used to elute the factor B oligomeric species from the column packed with DEAE-Sepharose Fast Flow resin. Finally, the purification procedure reported here refers to a typical experiment in which a bacterial mass derived from 1 liter of bacterial culture was used as the starting material for the recombinant bovine factor B preparation.
Analytical Ultracentrifugation
All sedimentation experiments were performed with a Beckman Optima XL-A ultracentrifuge at the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University of Texas Health Science Center, San Antonio. Data analyses were performed with UltraScan version 7.3 software [11]. Hydrodynamic corrections for buffer conditions were made according to data published previously [12], and as implemented in UltraScan. The partial specific volume of the recombinant bovine factor B was estimated from the polypeptide sequence according to the method of Durchschlag [13] using the values reported by Cohn and Edsall [14] as implemented in UltraScan and found to be 0.7389 ccm/g. Sedimentation equilibrium data were fit to multiple models, the most appropriate model was selected based on visual inspection of the residual run patterns and the fitting statistics. All samples were analyzed in a pH 8.0 buffer containing 50 mM Tris, 100 mM NaCl, 1 mM EDTA, and 1 mM TCEP. For equilibrium analysis, scans at equilibrium from multiple speeds (20,400; 27,000; 32,300; 36,900 and 40,600 rpm) were collected at 4°C, at both 291 nm and at 280 nm, in a radial step mode with a 0.001 cm step size setting and 20-point averages. Multiple loading concentrations ranging between 0.3-0.7 OD at each wavelength were measured; data exceeding 0.9 OD were excluded from the fit. Data in the concentration range between 0 and 37 μM were examined . For reversibly self-associating models, it is necessary to convert absorbance measurements into molar concentrations in order to fit for a global equilibrium constant. Since measurements were taken at multiple wavelengths, the different extinction coefficients at each wavelength must be considered. Extinction coefficients at 280 and 291 nm were determined by globally fitting absorbance wavelength scans as described previously [15], and were determined to be 31,160 and 20,420 M-1 cm-1, respectively. For the equilibrium experiments performed at 291 nm the maximum concentration used was 0.9 OD, which corresponds to 37 μM. Sedimentation velocity experiments were conducted at 20°C, 38,000 rpm and 293 nm, with a loading concentration of 43 μM. Experimental data were analyzed with the van Holde-Weischet analysis method [16, 17] to determine sedimentation coefficient distributions.
RESULTS
When a mixture of the cleavage products obtained following overnight treatment of the NusA-bovine factor B fusion polypeptide with protease thrombin was subjected to ion-exchange chromatography on DEAE-Sepharose Fast Flow in a 0-0.3 M NaCl gradient, two peaks with absorbance at 280 nm emerged from the column: a major peak eluted immediately following application of the sample to the resin, while a minor peak eluted with 0.05-0.1 M NaCl concentration (not shown; elution of the NusA moiety from the resin required a significantly higher salt concentration). SDS-PAGE analysis of the fractions constituting both peaks showed that they contained a highly pure recombinant bovine factor B, which exhibited a similar specific activity in an assay of ATP-driven NAD reduction by succinate catalyzed by reconstituted AE-SMP. The fractions constituting each peak were combined, concentrated and analyzed by the two separate size-sieving chromatographies on Sephacryl S-200 HR, the results of which are shown in Fig. 1. It is seen that the recombinant bovine factor B collected as the major peak from the preceding ion-exchange chromatography on DEAE-Sepharose eluted with a relative molecular weight of ∼56 kDa (fraction 43, Fig. 1A), while the recombinant protein detected in the minor peak eluted with a relative molecular weight of ∼34 kDa (fraction 47, Fig. 1B). Both peaks eluted from the Sephacryl S-200 HR column were broad and featured a characteristic right shoulder, indicative of a size heterogeneous population of the polypeptide. The 15% SDS-PAGE analysis demonstrated that each fraction contained a highly pure recombinant bovine factor B (see the Coomassie blue stained gels below the chromatograms shown in Fig. 1).
Figure 1.
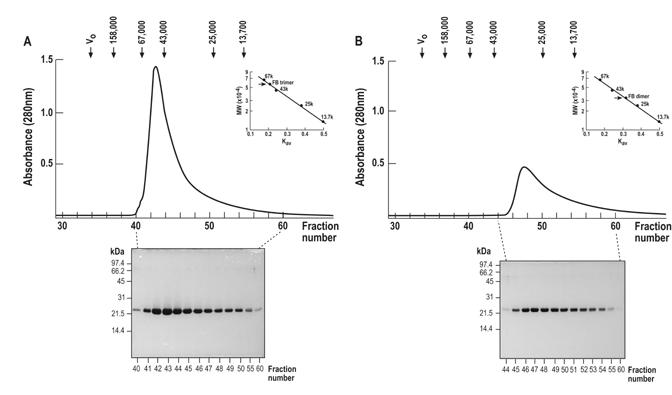
Size-sieving chromatography of recombinant bovine factor B on the Sephacryl S-200 HR column (1.5 x 95 cm). A, A major portion of the recombinant bovine factor B sample, obtained after overnight digestion of NusA-factor B fusion with thrombin, eluted at the beginning of the 0-0.3 M NaCl gradient from the ion-exchange chromatography on DEAE-Sepharose Fast Flow column; it was collected, concentrated and reapplied to the size-sieving Sephacryl S-200 HR. The elution profile was monitored by UV at 280 nm. The polypeptide composition of the fractions was analyzed by 15% SDS-PAGE followed by staining with Coomassie brilliant blue. The fractions with recombinant bovine factor B are shown. Inset shows the calibration curve of the column using the molecular weight standards. The molecular weight of factor B eluting in fraction 43 was estimated to be 56 kDa. B, A minor portion of the recombinant bovine factor B sample obtained from the thrombin digest of the NusA-factor B fusion eluted with 0.05-0.1 M NaCl concentration from the ion-exchange chromatography on DEAE-Sepharose Fast Flow column; it was collected, concentrated and reapplied to the size-sieving Sephacryl S-200 HR. The elution profile and the polypeptide composition of the fractions were analyzed as in panel A. Inset shows the calibration curve of the column using the molecular weight standards. The molecular weight of factor B eluting in fraction 47 was estimated to be 34 kDa. The molecular weight standards employed for calibration of the Sephacryl S-200 HR column were as follows: 2,000 kDa, blue dextran, 158 kDa, aldolase, 67 kDa, albumin, 43 kDa, ovalbumin, 25 kDa, chymotrypsinogen A, 13,7 kDa, ribonuclease A.
Fractions 40-48 shown in Fig. 1A and fractions 45-52 shown in Fig. 1B were each combined, concentrated to 10.5 and 2.4 mg protein per ml, respectively, and subjected to electrophoresis in the native conditions using the 7% Tris-acetate NuPAGE (Fig. 2). Staining of the native gel with Coomassie brilliant blue revealed a single protein species in lanes 1 and 2 (Fig. 2), suggesting a monodisperse distribution of the factor B molecules under experimental conditions. Incubation of the factor B preparations with 6 M urea led to a disappearance of the polypeptide from the native gel (not shown).
Figure 2.
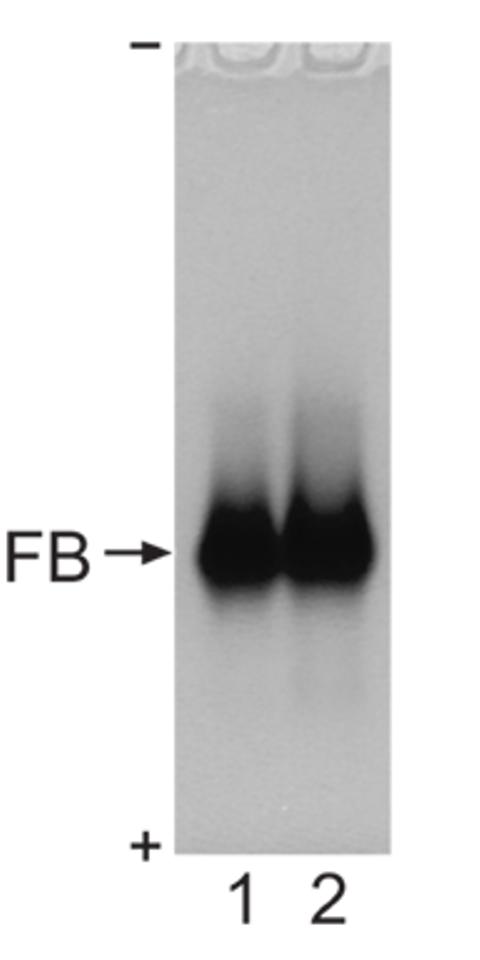
Analysis of recombinant bovine factor B by a native polyacrylamide gel. Lane 1, 21 μg of factor B from combined fractions 40-48 of the size-sieving chromatography shown in Fig. 1A, at 10.5 mg protein per ml, and lane 2, 24 μg of factor B from combined fractions 45-52 of the size-sieving chromatography shown in Fig. 1B at 2.4 mg protein per ml, were analyzed by 7% Tris-Acetate NuPAGE (Invitrogen), and stained with Coomassie brilliant blue. (-) indicates the position of the cathode, and (+) indicates the position of the anode. In the native electrophoresis gel system, the recombinant bovine factor B species migrated from the top to the bottom.
To probe the quaternary structure of the recombinant bovine factor B in solution, we employed a chemical cross-linking approach. We used three different cross-linkers that varied in the chemical nature of their reactive group or the length of the bridging them spacer: 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), a zero-length cross-linker that reacts with carboxyl groups, in the presence of N-hydroxysulfosuccinimide, to enhance the coupling reaction at physiological pH; disulfosuccinimidyl tartrate (sulfo-DST), with a spacer arm that is 6.4 Å in length; and bis (sulfosuccinimidyl) suberate (BS), with a spacer arm that is 11.4 Å. Both sulfo-DST and BS react with amino groups. No cross-linked products were detected after treating the recombinant factor B with EDC in the presence of N-hydroxysulfosuccinimide; treatment with sulfo-DST resulted in low yields of a cross-linked product with Mr of ∼43 kDa (Fig. 3A, lanes 5 and 6). A distinct cross-linking pattern was observed after reacting the recombinant bovine factor B (at concentration of 1.5 mg protein per ml) with 1 mM BS for 5 and 10 min (Fig. 3A, lanes 3 and 4).
Figure 3.
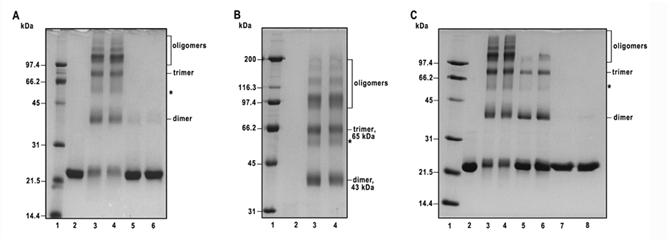
Probing of the oligomeric structure of recombinant bovine factor B by chemical cross-linking. A, The cross-linking pattern of recombinant factor B obtained using bis (sulfosuccinimidyl) suberate (BS, spacer arm 11. 4 Å) and disulfosuccinimidyl tartrate (sulfo-DST, spacer arm 6.4 Å). Lane 2, control, no cross-linker added; lanes 3 and 4, cross-linking of factor B (1.5 mg protein per ml) with 1 mM BS for 5 min and 10 min at room temperature, respectively; lanes 5 and 6, cross-linking of factor B (1.5 mg protein per ml) with 1 mM sulfo-DST for 5 and 10 min, respectively. The cross-linking reactions were quenched with 100 mM ammonium acetate, and the samples were analyzed by 12% SDS-PAGE under reducing conditions, followed by staining with Coomassie brilliant blue. Each lane was loaded with 4 μg of factor B. B, Samples identical to those shown in lanes 2-4 of A were separated on 9 % SDS-PAGE under reducing conditions. In the experimental conditions employed, the non-cross-linked factor B ∼23 kDa monomeric species ran off the gel and is not visible in the lane 2. Lanes 3 and 4 contain recombinant factor B cross-linked with 1 mM BS for 5 and 10 min, respectively. Each lane was loaded with 6.5 μg of factor B. C, The effect of factor B protein concentration on cross-linking pattern with 1 mM BS. Lane 2, control, no cross-linker added; lanes 3 and 4, factor B (2 mg protein per ml) cross-linked with 1 mM BS for 5 and 10 min, respectively; lanes 5 and 6, factor B (0.2 mg protein per ml) cross-linked with 0.1 mM BS for 5 and 10 min, respectively; lanes 7 and 8, factor B (0.02 mg protein per ml) cross-linked with 10 μM BS for 5 and 10 min, respectively. Each lane was loaded with 5.2 μg of factor B. Shown is a 12% SDS-PAGE stained with Coomassie brilliant blue. Lane 1 in A and C, and lane 1 in B contain low and high molecular weight standards, respectively, and their Mr values (in kDa) are shown on the left.
To determine the relative molecular weights of the cross-linked products accurately, the samples shown in lanes 2-4 of Fig. 3A were separated using 9% SDS-PAGE (Fig. 3B, lanes 2-4). At this concentration of acrylamide, the non-cross-linked factor B, which migrates in 15% SDS-PAGE with an Mr of ∼23 kDa, ran off the gel, and is not present in lane 2 of Fig. 3B. The most prominent products generated from the reaction of BS with factor B included bands with Mr,s of ∼43 and ∼65 kDa, as well as higher molecular weight aggregates labeled as “oligomers” in Fig. 3. A low yield cross-linked product with an Mr of ∼ 55 kDa was observed, which may represent an alternative pathway of the factor B cross-linking; it is marked with an asterisk in Fig. 3.
The formation of the cross-linked products with Mrs of ∼43 and ∼65 kDa, as well as the higher molecular weight oligomers was a function of factor B concentration (Fig. 3C). In the experiment shown in Figure 3C, the molar ratio of the total amino groups (including the ε-amino groups of factor B 14 Lys residues plus its NH2-terminal group) to BS concentration was kept constant, at a 1.5:1 ratio, while the factor B protein concentration varied 100-fold, from 2 to 0.02 mg protein per ml (Fig. 3C, lanes 3 and 4, and lanes 7 and 8, respectively). Lowering the protein concentration from 2 to 0.2 mg protein per ml most dramatically affected the yield of the oligomers of Mr ∼100 kDa and higher. A significant decrease in the yield of the band with an Mr of ∼65 kDa, labeled as a trimer in Fig. 3, is also noticeable. No cross-linked products were detected when factor B, at a protein concentration of 0.02 mg per ml, was cross-linked with 10 μM BS for 5 or 10 min (Fig. 3C, lanes 7 and 8).
We next analyzed the formation of the cross-linked products while maintaining the factor B concentration at 1.6 mg per ml, and varying the cross-linker concentration within a range of 0.025-1.0 mM (Fig. 4). Employing BS concentrations of 0.025, 0.05 or 0.1 mM favored the formation of cross-linked products with Mr values of ∼43 and ∼65 kDa (see labels “dimer” and ‘trimer” in Fig. 4). The yield of the dimer and trimer species increased with increasing cross-linker concentration until their peak yields were reached, which occurred at BS concentrations of 0.1 and 0.4 mM, respectively. At cross-linker concentrations greater then those that produced the maximal yields, the dimer and trimer species yields declined, while the oligomer yields continued to increase. The cross-linking results presented in Fig. 4 suggest that formation of the factor B dimer and trimer cross-linked species at low BS concentrations represents an initial step of the chemical reaction, and reflects an existence of factor B monomer-trimer equilibrium in solution. Subsequent spurious modification of the protein’s Lys residues at higher BS concentrations leads to protein unfolding and the appearance of the non-specific high molecular weight aggregates.
Figure 4.
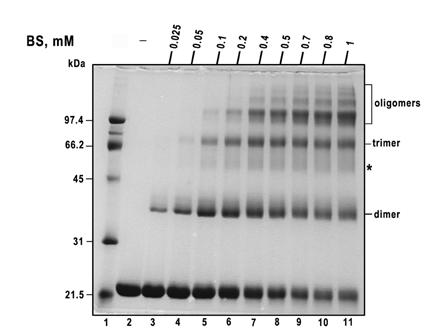
Effect of the BS concentration on the factor B cross-linking pattern. Recombinant factor B (1.6 mg protein per ml) was incubated at room temperature with the indicated concentrations of BS. Following a 5 min incubation with the cross-linker addition, the reaction was quenched with 100 mM ammonium acetate. Lane 2, control, no cross-linker added; lane 3, 0.025 mM BS; lane 4, 0.05 mM BS; lane 5, 0.1 mM BS; lane 6, 0.2 mM BS; lane 7, 0.4 mM BS; lane 8, 0.5 mM BS; lane 9, 0.7 mM BS; lane 10, 0.8 mM BS; lane 11, 1 mM BS. Each lane was loaded with ∼ 8.2 μg of factor B. Shown is a 10% SDS-PAGE stained with Coomassie brilliant blue. Lane 1 contains low molecular weight standards and their Mr values (in kDa) are shown on the left.
We then analyzed the oligomerization state of bovine factor B using sedimentation velocity and sedimentation equilibrium analyses. Van Holde-Weischet analysis of the velocity data revealed a sedimentation distribution ranging between 1.7 s and 3.85 s, with a weight average sedimentation coefficient of 3.32 (Fig. 5). The sedimentation coefficient distribution displayed the half-parabola-shaped pattern characteristic of a reversibly, self-associating system as discussed previously [18]. The range of sedimentation coefficients suggests that the largest particle is more than twice as large as the smallest particle in the distribution, supporting a monomer-trimer model.
Figure 5.
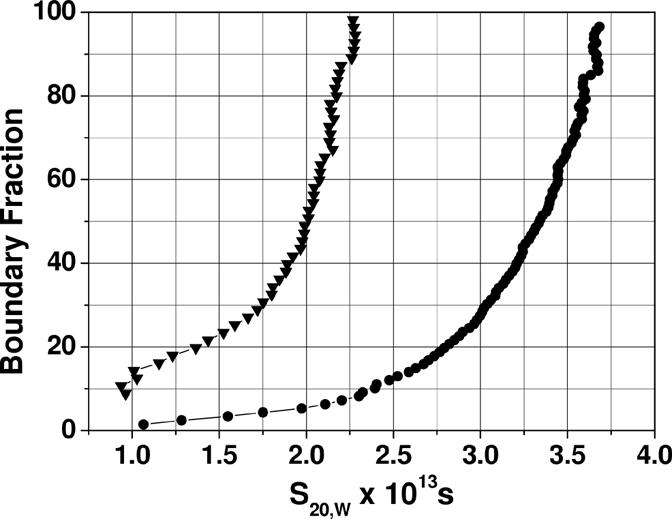
Van Holde-Weischet integral distribution plot for bovine factor B. Shown are distributions for two different loading concentrations (filled triangles = 5 μM, filled circles = 40 μM). The distribution displays the characteristic half-parabola shaped distribution for a reversibly self-associating system, and shows a clear shift towards s20,W-values consistent with a reversible dissociation upon dilution. The range of sedimentation coefficients observed indicates a stoichiometry greater than 1:2 for the association.
To further determine the oligomerization properties of bovine factor B, we performed sedimentation equilibrium experiments. By globally fitting data observed under multiple conditions used in the sedimentation equilibrium experiments, such as multiple rotor speeds and multiple loading concentrations, it is possible to enhance the confidence in each fitted parameter value [19]. In such a fit, parameters such as monomer molecular weight are considered global and are forced to be the same for all included datasets. In order to interpret the data correctly, results from all possible models should be considered and compared. In general, smaller variances result in a better fit. However, it is important to use the model that provides the lowest variance and the fewest adjustable parameters. In this case, the lowest variance was obtained with a two-component, non-interacting ideal species model, which does not place any global constraints on the molecular weights of each species or the relative protein species concentrations (Table 1), other than to require that both molecular weight terms are identical for all measured scan conditions. Here the RMSD values were close or below the intrinsic noise. The molecular weights reported by this model are in excellent agreement with those calculated for the monomer and trimer of bovine factor B. The next best fit was obtained by the monomer-trimer and the monomer-dimer-trimer models, which both had the same variance, only 1.2 fold higher than the variance reported for the two-component noninteracting model. Although these models allow for two or three components, a constraint forces any additional components to be an integer multiple of the first, and the concentration of each protein species is constrained by a globally fitted equilibrium constant, permitting a reduction in the number of fitting parameters from 63 to 42 and 43, respectively. From these results it can be seen that both models report monomer molecular weights very close to that of the bovine factor B monomer. However, the variance is not improved by adding a dimer to the model, and the monomer-dimer dissociation constant in the monomer-dimer-trimer model is out of range of the measurements, indicating that no confidence can be placed in this parameter, and that it is not necessary to describe the data. In addition, the 95% confidence intervals, as determined by Monte Carlo analysis, for the monomer molecular weight and the dissociation constant are larger for the monomer-dimer-trimer model than those for the values of the monomer-trimer model. Using 41 floating parameters, a single ideal species model reports a variance that is almost twice as high as the variance for the monomer-trimer model, and therefore has to be excluded. Similarly, a monomer-dimer model displayed a 25% higher variance than the monomer-trimer model, with the same number of fitting parameters. The monomer-dimer model also resulted in a monomer molecular weight inconsistent with any integral multiple of the known molecular weight of bovine factor B. Taken together, we conclude from these results that bovine factor B assembles in a monomer-trimer, reversible self-association process without any significant amount of dimer intermediate, with a dissociation constant Kd1,3 = 2.48 × 10-10M2. The monomer-trimer fit is shown in Fig. 6, and a distribution of the monomer and trimer species is shown in Fig. 7.
Table 1.
Sedimentation equilibrium results for studies on bovine factor B. According to the variance, number of floated parameters, and monomer molecular weights reported, these results show that the monomer-trimer model is the most appropriate model choice. The 95% confidence intervals determined by Monte Carlo analysis are reported in parentheses.
| Equilibrium Model | Molecular weight | RMS (x 102) | Kd | Floated Parameters |
|---|---|---|---|---|
| single ideal species | 42.14 kDa (+.38/-.35) | 2.380 | - | 41 |
| monomer-dimer | 29.29 kDa (+2.07/-3.31) | 1.997 | Kd1,2 = 2.57 × 10-5 M (+3.08/-1.40 x 10-5M) | 42 |
| monomer-trimer | 20.97 kDa (+1.51/-1.08) | 1.755 | Kd1,3 = 2.48 × 10-10 M2 (+1.42/-0.90 × 10-10 M2) | 42 |
| monomer-dimer-trimer | 21.04 kDa (+1.44/-1.22) | 1.755 | Kd1,2: not detectable: Kd1,3 = 2.56 × 10-10 M2 (+1.51/-0.95 × 10-10 M2) | 43 |
| Two ideal species,non-interacting | 22.61 kDa (+.73/-.68) 60.84 kDa (+2.46/-1.84) | 1.595 | - | 63 |
| Calculated from Protein Sequence | 20.42 kDa | (monomer) |
Figure 6.
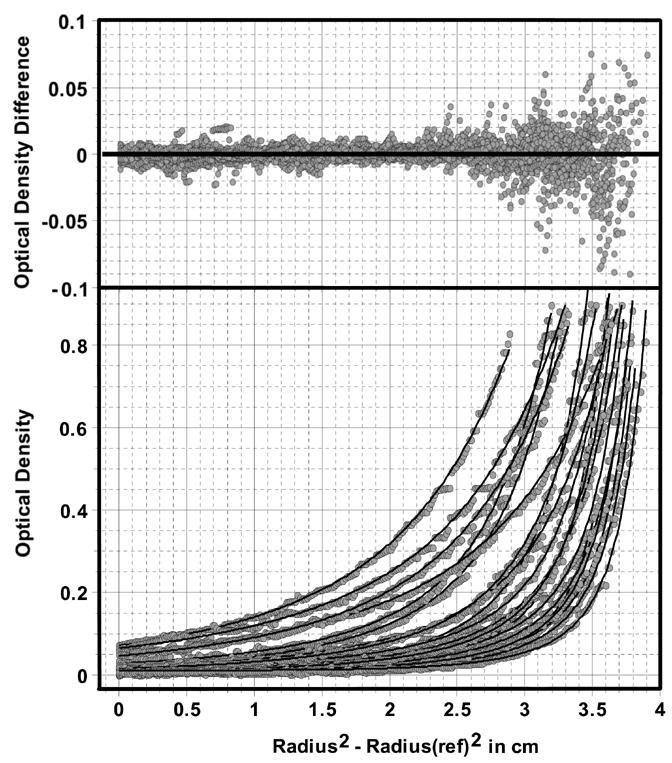
Global sedimentation equilibrium analysis results for recombinant bovine factor B.Shown is the fit to a monomer-trimer model for five speeds and four loading concentrations measured at 280 nm and 291 nm. Fitted overlays (solid lines) to experimental data (grey circles) are shown in the bottom panel; residuals are shown in the upper panel. The trimer dissociation constant (Kd1,3 = [molar concentration of monomer]3/[molar concentration of trimer]) was found to be 2.48 × 10-10 M2.
Figure 7.
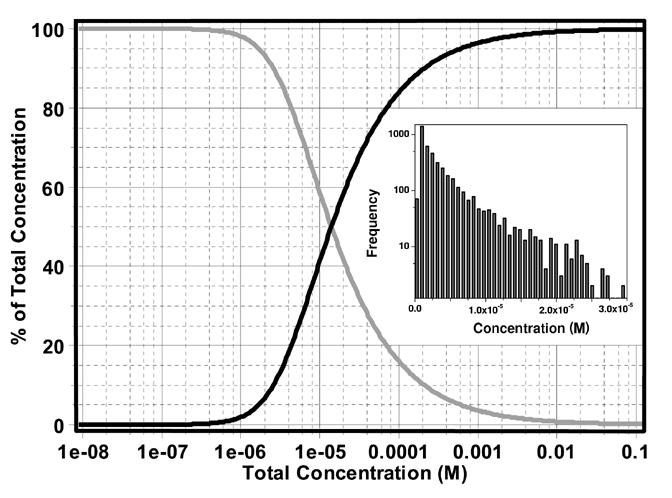
Distribution of monomer (grey line) and trimer (black line) concentration vs. total concentration of recombinant bovine factor B. As shown in this plot, the dissociation constant is well described by the range of the measured concentrations. Therefore, the data provide signal for both the monomeric and trimeric factor B species. The inset shows the relative frequencies of data points for each concentration.
DISCUSSION
The major result of the current study is that the recombinant bovine factor B exhibits a reversible self-associating behavior in solution that is best described by a monomer-trimer equilibrium model. Five lines of evidence support this conclusion. First, using size-sieving chromatography, a distribution of the factor B species between a monomer and a trimer, but no oligomers of higher molecular weight, were observed. Second, at high protein concentrations, factor B migrated as a single band in the native 7% Tris-acetate gel at pH 7.0. Third, cross-linking of factor B with an amino-reactive cross-linking reagent revealed that at low cross-linker-protein ratio the primary cross-linked products were the factor B dimer and trimer. Fourth, in a sedimentation velocity experiment, the range of sedimentation coefficients suggested that the largest particle present in the distribution was more than twice as large as the smallest. While we did not specifically investigate the frictional ratios of monomer and trimer, we do note that the s-value ratio of maximum over minimum is larger than 2.0. Since the shape of the van Holde-Weischet s20,W-value distribution clearly indicates a pattern consistent with a rapidly reversible association, the limits of the distribution do not necessarily reflect the exact s20,W-values of the limiting components (i.e., monomer and trimer) and instead show s20,W-values from the reaction boundary. Furthermore, the van Holde-Weischet analysis does not explicitly yield molecular weights, frictional-or diffusion coefficients. Taking the smallest observed s20,W-value for the monomer (1.7s) and the largest value from the distribution (3.85s) for the trimer, the partial specific volume of factor B (.7389 ccm/g) and the molecular weight of monomer (20,420 Da) and trimer (61,260 Da) we calculate a frictional ratio of ∼1.5 for the monomer and ∼1.4 for the trimer. An f/f0 value of 1.5 for the monomer suggests an elongated structure of the monomer. A reduction of the frictional coefficient indicates that the association produces a more globular structure, suggesting a side-to-side association, rather than an end-to-end association. We cannot unambiguously assign this value to the trimer, most likely the observed maximum is somewhat lower than the actual trimer s20,W-value, which would also make the frictional coefficient slightly lower than 1.4. Finally, the data obtained under multiple conditions in the sedimentation equilibrium experiments were found to fit best to a model describing a reversible self association of a monomer-trimer of factor B, with a monomer-trimer dissociation constant Kd1,3 = 2.48 × 10-10 M2.
The structural basis underlying factor B oligomerizaton is not clear. It could be related to a recently identified weak match of the cysteine-containing leucine-rich repeat (LRR) motif to the factor B sequence comprising amino acids 62-145 [5]. The LRR motif is 20-29 residues long and contains a conserved 11-residue consensus sequence LxxLxLxxN/CxL, in which x represents any amino acid and L positions can also be occupied by valine, isoleucine or phenylalanine. The motif is characterized by an α/β fold with intervening loops, and in the original family member ribonuclease inhibitor, 15 LRRs are arranged to form a “horseshoe” structure [20]. The LRR motif has been identified in a variety of proteins, and has been implicated in protein-protein interactions [21]. Recent experiments with a 10.5 kDa proteolytic fragment of bovine factor B, encompassing its LRR-like sequence, demonstrated that this fragment formed dimeric and trimeric species following cross-linking with BS (G. I. B., unpublished). This observation suggests that the LRR-like motif in factor B could be considered as a putative oligomerization domain.
Several mitochondrial proteins of varied molecular weights, including oligomers comprised of lower molecular weight monomers, each claimed to possess a factor B-like coupling activity, were described in the early bioenergetics literature [1]. Although no direct comparison is currently possible between these preparations derived from bovine heart mitochondria and our recombinant bovine factor B, reports from the Sanadi [2, 22] and Wang [23] laboratories describing the coupling factor B species with molecular weights of 32 [2], 47 [22] or 330 kDa [23] could be considered as early evidence of factor B tendency for oligomerization.
The physiological significance of the recombinant bovine factor B oligomerization described in the present study has yet to be determined. Analysis of stimulation of the rate of ATP-driven NAD reduction by succinate catalyzed by AE-SMP reconstituted with increasing amounts of the recombinant human factor B suggests that the polypeptide binds to the mitochondrial inner membrane in quantities that are nearly stoichiometric to that of the ATP synthase complex concentration in submitochondrial particles [3]. The content of the latter enzyme in the mitochondrial inner membrane is estimated to be within 0.4-0.45 nmole per mg of membrane protein [24]. Given the value of 0.6 μl per mg of protein as an approximation for the volume of the mitochondrial matrix [25], the concentration of a pool of soluble factor B imported into the mitochondria could be within the equilibrium concentration range of 16 μM that was found in the present study. It remains to be seen, however, whether self-association of recombinant bovine factor B described here affects the behavior of the polypeptide in vivo. Regardless of recombinant bovine factor B’s oligomerization properties, the concentrations of the polypeptide required for attaining its maximal coupling activity in the activity assays [3-5] are below 1 μM, and thus, the functionally active factor B species is a monomer.
The mammalian ATP synthase complex is comprised of 17 distinct polypeptides [3, 26]. These polypeptides are present in varying stoichiometries and are assembled into two major sectors, F1, which catalyzes ATP synthesis and hydrolysis, and F0, which is involved in proton translocation across the membrane, and which are linked by central and peripheral stalks. Among its 17 polypeptides, the ATPase inhibitor protein IF1 and factor B have been assigned regulatory roles and seem to function in concert, forming an intricate two tiered-system that protects cells from a sudden loss of the two major forms by which a eukaryotic cell stores its energy, ATP and Δμ +H. IF1 binds to the F1 β subunit and prevents futile ATP hydrolysis under conditions of low proton-motive force [27]. In contrast, under physiological conditions of oxidative phosphorylation, factor B interacts with an as yet unidentified component of membrane sector F0 to facilitate ATP synthesis by blocking a futile proton leak across the mitochondrial inner membrane [3-5]. We recently hypothesized that under conditions of high levels of a proton-motive force (i.e. during a low work load or state 4 respiration, when the rate of the reactive oxygen species (ROS) production is elevated), factor B may function as a pressure valve for maintaining the proton-motive force below a critical threshold, by mildly uncoupling the ATP synthase complex [3]. The underlying mechanism of such an action could involve a factor B conformation change caused by reversible oxidation of its vicinal dithiol. Under pathological conditions, characterized by excessive ROS production and a failure of the intra-mitochondrial antioxidant network [28], binding of the protein inhibitor IF1 to F1 in a ΔμH +-dependent manner [29] could spare the cell from a precipitous loss of intracellular ATP.
A decade ago we described the membrane topology and near-neighbor relationships of subunits e, f and g of the membrane sector F0 of the bovine heart mitochondria ATP synthase complex [30]. Specifically, we demonstrated that subunit e, with a calculated molecular mass of 8.2 kDa, is anchored to the mitochondrial inner membrane via a hydrophobic NH2-terminal segment, while its hydrophilic, coiled-coil COOH-terminal half is exposed into the mitochondrial intermembrane space. Using chemical cross-linkers of varying reactivities, we demonstrated formation of the cross-linked products identified as e-e or e-g dimers. These observations were extended subsequently by the studies from the Stuart [31, 32] and Velours [33-35] laboratories that demonstrated that yeast counterparts of the bovine subunits e and g are required for the oligomerization of the yeast ATP synthase, and that inactivation of the genes encoding these polypeptides perturbs the regular structure of the mitochondrial cristae. Thus, ATP synthase oligomerization is envisioned to be mediated by protein-protein interactions involving the intramembraneous and extramembraneous segments of the F0 subunits, protruding into the intermembrane space. A recent crystal structure of F1 dimers, linked together via the antiparallel COOH-terminal coiled-coil domain of the inhibitor protein IF1 [36], suggests that there may be an additional mechanism by which ATP synthase dimerization is promoted that may rely on protein-protein interactions between components of the enzyme that are located on the matrix-facing side of the mitochondrial inner membrane. However, dimerization of the bovine [37] or yeast [38] mitochondria ATP synthase complex was shown to be independent of the inhibitor protein IF1. Whether factor B has a role in ATP synthase complex oligomerization remains to be investigated.
ACKNOWLEDGEMENTS
G.I.B. thanks Dr. Youssef Hatefi (retired) for continuing support, encouragement, and inspiration; Dr. George Sachs for hospitality; Dr. Keith Munson for enlightening discussions and his suggestion to perform the experiment shown in Figure 4; and Dr. Martin Phillips for participation in an early stage of this study.
Glossary
The abbreviations used are:
- AE-SMP
SMP depleted of factor B
- SMP
bovine heart submitochondrial particles
- FB
a regulatory component of ATP synthase complex factor B/subunit s
- SDS-PAGE
sodium dodecylsulfate polyacrylamide gel electrophoresis
- DTT
dithiothreitol
- TCEP
Tris (2-carboxyethylphosphine) hydrochloride
- EDTA
ethylenediaminetetraacetic acid
- Tris
Tris (hydroxymethyl) aminomethane
- IPTG
isopropyl-β-D-thiogalactopyranoside)
- BS
Bis (sulfosuccinimidyl) suberate
- sulfo-DST
(Disulfosuccinimidyl tartrate)
- EDC
1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride.
Footnotes
This work was supported by NIH Grant GM66085 to G.I.B. The development of the UltraScan software is in part supported by the NSF through grant DBI-9974819 to B. D. and by grant # 10001642 from the San Antonio Life Science Institute to B. D.
REFERENCES
- [1].Sanadi DR. Mitochondrial coupling factor B. Properties and role in ATP synthesis. Biochim. Biophys. Acta. 1982;683:39–56. [Google Scholar]
- [2].Lam KW, Warshaw JB, Sanadi DR. The mechanism of oxidative phosphorylation. XIV. Purification and properties of a second energy-transfer factor. Arch. Biochem. Biophys. 1967;119:477–484. doi: 10.1016/0003-9861(67)90479-1. [DOI] [PubMed] [Google Scholar]
- [3].Belogrudov GI, Hatefi Y. Factor B and the mitochondrial ATP synthase complex. J. Biol. Chem. 2002;277:6097–6103. doi: 10.1074/jbc.M111256200. [DOI] [PubMed] [Google Scholar]
- [4].Belogrudov GI. Factor B is essential for ATP synthesis by mitochondria. Arch. Biochem. Bio-phys. 2002;406:271–274. doi: 10.1016/s0003-9861(02)00431-9. [DOI] [PubMed] [Google Scholar]
- [5].Belogrudov GI. Bovine factor B: Cloning, expression, and characterization. Arch. Biochem. Biophys. 2006;451:68–78. doi: 10.1016/j.abb.2006.02.021. [DOI] [PubMed] [Google Scholar]
- [6].Lee C-P, Ernster L. Restoration of oxidative phosphorylation in non-phosphorylating submi-tochondrial particles by oligomycin. Biochem. Biophys. Res. Commun. 1965;18:523–529. doi: 10.1016/0006-291x(65)90785-0. [DOI] [PubMed] [Google Scholar]
- [7].Ausubel FM, Brent R, Kingston RE, et al., editors. Current Protocols in Molecular Biology. Wiley Interscience; New York: 1997. [Google Scholar]
- [8].Joshi S, Sanadi DR. Purification of coupling factor B. Methods Enzymol. 1979;55:384–391. doi: 10.1016/0076-6879(79)55050-2. [DOI] [PubMed] [Google Scholar]
- [9].Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Analytical Biochemistry. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- [10].Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- [11].Demeler B. University of Texas Health Science Center at San Antonio, Dept. of Biochemistry. http://www.ultrascan.uthscsa.edu.
- [12].Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. Analytical Ultracentrifugation in Biochemistry and Polymer Science. Royal Society of Chemistry; Cambridge: 1992. pp. 90–125. [Google Scholar]
- [13].Durchschlag H. Specific volumes of biological macromolecules and some other molecules of biological interest. In: Hinz H-J, editor. Thermodynamic Data for Biochemistry and Biotechnology. Springer-Verlag; New York: 1986. pp. 45–128. [Google Scholar]
- [14].Cohn EJ, Edsall JT. Proteins, amino acids, and peptides as ions and dipolar ions. Reinhold; New York: New York: 1965. reprint by Hafner. [Google Scholar]
- [15].Demeler B, Scott DJ, Harding SE, Rowe AJ. Modern Analytical Ultracentrifugation: Techniques and Methods. Royal Society of Chemistry; Cambridge: 2005. A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments; pp. 210–229. [Google Scholar]
- [16].van Holde KE, Weischet WO. Boundary Analysis of Sedimentation-Velocity Experiments with Monodisperse and Paucidisperse Solutes. Biopolymers. 1978;17:1387–1403. [Google Scholar]
- [17].Demeler B, van Holde KE. Sedimentation velocity analysis of highly heterogeneous systems. Anal. Biochem. 2004;335:279–288. doi: 10.1016/j.ab.2004.08.039. [DOI] [PubMed] [Google Scholar]
- [18].Demeler B, Saber H, Hansen JC. Identification and interpretation of complexity in sedimentation velocity boundaries. Biophys. J. 1997;72:397–407. doi: 10.1016/S0006-3495(97)78680-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Johnson ML, Correia JJ, Yphantis DA, Halvorson HR. Analysis of data from the analytical ultracentrifuge by nonlinear least-squares techniques. Biophys. J. 1981;36:575–588. doi: 10.1016/S0006-3495(81)84753-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kobe B, Deisenhofer J. Crystal structure of porcine ribonuclease inhibitor, a protein with leucine-rich repeats. Nature. 1993;366:751–756. doi: 10.1038/366751a0. [DOI] [PubMed] [Google Scholar]
- [21].Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Current Opinion Struc. Biol. 2001:725–732. doi: 10.1016/s0959-440x(01)00266-4. [DOI] [PubMed] [Google Scholar]
- [22].Shankaran R, Sani BP, Sanadi DR. Studies on oxidative phosphorylation: evidence for multiple forms of factor B activity. Arch. Biochem. Biophys. 1975;168:394–402. doi: 10.1016/0003-9861(75)90268-4. [DOI] [PubMed] [Google Scholar]
- [23].Higashiyama T, Steinmeier RC, Serianne BC, Knoll SL, Wang JH. Isolation and characterization of coupling factor FB from bovine heart mitochondria. Biochemistry. 1975;14:4117–4121. [Google Scholar]
- [24].Matsuno-Yagi A, Hatefi Y. Estimation of the turnover number of bovine heart F0F1 complexes for ATP synthesis. Biochemistry. 1988;27:335–340. doi: 10.1021/bi00401a050. [DOI] [PubMed] [Google Scholar]
- [25].Brand MD. Measurement of mitochondrial protonmotive force. In: Brown GC, Cooper CE, editors. Bioenergetics: a Practical Approach. IRL Press at Oxford University Press; Oxford: 1995. pp. 39–62. [Google Scholar]
- [26].Ko YH, Delannoy M, Hullihen J, Chiu W, Pedersen PL. Mitochondrial ATP synthasome. Cristae-enriched membranes and a multiwell detergent screening assay yield dispersed single complexes containing the ATP synthase and carriers for Pi and ADP/ATP. J. Biol. Chem. 2003;278:12305–12309. doi: 10.1074/jbc.C200703200. [DOI] [PubMed] [Google Scholar]
- [27].Schwerzmann K, Pedersen PL. Regulation of the mitochondrial ATP synthase/ATPase complex. Arch. Biochem. Biophys. 1986;250:1–18. doi: 10.1016/0003-9861(86)90695-8. [DOI] [PubMed] [Google Scholar]
- [28].Cadenas E. Mitochondrial free radical production and cell signaling. Mol. Aspects Med. 2004;25:17–26. doi: 10.1016/j.mam.2004.02.005. [DOI] [PubMed] [Google Scholar]
- [29].Green DW, Grover GJ. The IF1 inhibitor protein of the mitochondrial F1F0-ATPase. Biochim Biophys. Acta. 2000;1458:343–355. doi: 10.1016/s0005-2728(00)00085-2. [DOI] [PubMed] [Google Scholar]
- [30].Belogrudov GI, Tomich JM, Hatefi Y. Membrane topography and near-neighbor relationships of the mitochondrial ATP synthase subunits e, f, and g. J. Biol. Chem. 1996;271:20340–20345. doi: 10.1074/jbc.271.34.20340. [DOI] [PubMed] [Google Scholar]
- [31].Arnold I, Pfeiffer K, Neupert W, Stuart RA, Schagger H. Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits. EMBO J. 1998;17:7170–7178. doi: 10.1093/emboj/17.24.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Saddar S, Stuart RA. The yeast F1F0-ATP synthase: analysis of the molecular organization of subunit g and the importance of a conserved GXXXG motif. J. Biol. Chem. 2005;280:24435–24442. doi: 10.1074/jbc.M502804200. [DOI] [PubMed] [Google Scholar]
- [33].Giraud MF, Paumard P, Soubannier V, Vaillier J, Arselin G, Salin B, Schaeffer J, Brethes D, di Rago JP, Velours J. Is there a relationship between the supramolecular organization of the mitochondrial ATP synthase and the formation of cristae? Biochim. Biophys. Acta. 2002;1555:174–180. doi: 10.1016/s0005-2728(02)00274-8. [DOI] [PubMed] [Google Scholar]
- [34].Arselin G, Vaillier J, Salin B, Schaeffer J, Giraud MF, Dautant A, Brethes D, Velours J. The modulation in subunits e and g amounts of yeast ATP synthase modifies mitochondrial cristae morphology. J. Biol. Chem. 2004;279:40392–40399. doi: 10.1074/jbc.M404316200. [DOI] [PubMed] [Google Scholar]
- [35].Bustos DM, Velours J. The modification of the conserved GXXXG motif of the membranespanning segment of subunit g destabilizes the supramolecular species of yeast ATP synthase. J. Biol. Chem. 2005;280:29004–29010. doi: 10.1074/jbc.M502140200. [DOI] [PubMed] [Google Scholar]
- [36].Cabezon E, Montgomery MG, Leslie AG, Walker JE. The structure of bovine F1-ATPase in complex with its regulatory protein IF1. Nat. Struct. Biol. 2003;10:744–750. doi: 10.1038/nsb966. [DOI] [PubMed] [Google Scholar]
- [37].Tomasetig L, Di Pancrazio F, Harris DA, Mavelli I, Lippe G. Dimerization of F0F1-ATP synthase from bovine heart is independent from the binding of the inhibitor protein IF1. Biochim. Biophys. Acta. 2002;1556:133–141. doi: 10.1016/s0005-2728(02)00344-4. [DOI] [PubMed] [Google Scholar]
- [38].Dienhart M, Pfeiffer K, Schagger H, Stuart RA. Formation of the yeast F1F0-ATP synthase dimeric complex does not require the ATPase inhibitor protein, Inh1. J. Biol. Chem. 2002;277:39289–30295. doi: 10.1074/jbc.M205720200. [DOI] [PubMed] [Google Scholar]