

Abstract
Background
Allopolyploidy is a preeminent process in plant evolution that results from the merger of distinct genomes in a common nucleus via inter-specific hybridization. Allopolyploid formation is usually related to genome-wide structural and functional changes though the underlying mechanisms operating during this "genomic shock" still remain poorly known. The aim of the present study was to investigate the modifications occurring at the proteomic level following an allopolyploidization event and to determine whether these changes are related to functional properties of the proteins. In a previous report, we applied comparative proteomics to synthetic amphiploids of Brassica napus and to its diploid progenitors B. rapa and B. oleracea. Although several hundred polypeptides displayed additivity (i.e. mid-parent values) in the amphiploids, many of them showed non-additivity. Here, we report the in silico functional characterization of the "non-additive" proteins (the ones with a non-additive pattern of regulation) in synthetic B. napus.
Results
The complete set of non-additive proteins (335 in the stem and 205 in the root), as well as a subset of additive polypeptides (200 per organ), was identified by mass spectrometry. Several protein isoforms were found, and most of them (~55%) displayed "different" or "opposite" patterns of regulation in the amphiploids, i.e. isoforms of the same protein showing both up-regulation and down-regulation in the synthetic B. napus compared to the mid-parent value. Components of protein complexes were identified of which ~50% also displayed "different" or "opposite" patterns of regulation in the allotetraploids. In silico functional categorization of the identified proteins was carried out, and showed that neither functional category nor metabolic pathway were systematically affected by non-additivity in the synthetic amphiploids. In addition, no subcellular compartment was found to be over- or under-represented among the proteins displaying non-additive values in the allopolyploids.
Conclusion
Protein identification showed that functionally related polypeptides (isoforms and complex subunits) could be differentially regulated in synthetic B. napus in comparison to its diploid progenitors while such proteins are usually expected to display co-regulation. The genetic redundancy within an allopolyploid could explain why functionally related proteins could display imbalanced levels of expression. No functional category, no metabolic pathway and no subcellular localization was found to be over- or under-represented within non-additive polypeptides, suggesting that the differential regulation of gene products was not related to functional properties of the proteins. Thus, at the protein level, there is no evidence for the "genomic shock" expected in neo-polyploids and the overall topology of protein networks and metabolic pathways is conserved in synthetic allotetraploids of B. napus in comparison to its diploid progenitors B. rapa and B. oleracea.
Background
Polyploidization is a widespread phenomenon in eukaryotes, especially in plants where most or all angiosperms are current polyploids or have polyploid origins [1-3]. The last few years have witnessed new insights into the evolutionary and ecological significance of both auto- and allo-polyploidy in plants [4-8], and these attributes have been related to structural and functional genomic changes [9-12]. In particular, the analyses of synthetic allopolyploids have revealed extensive modifications in gene expression. According to the "additivity" hypothesis, newly-synthesized allopolyploids are supposed to display mid-parent expression patterns. Many exceptions were found in synthetic allopolyploids (e.g. Arabidopsis [11], Senecio [13], Brassica [14], Triticum [15], and Gossypium neo-polyploids [16]), suggesting that the differential regulation of gene expression is a common feature of plant allopolyploids. Although the phenomenon of non-additive expression in inter-specific hybrids and allopolyploids is now well described, the underlying mechanisms are still poorly understood. Several approaches may be developed to decipher the mechanisms of gene regulation in polyploids. The investigation of candidate causal processes, such as epigenetic mechanisms or RNA inactivation [17,18], may give valuable information. Another main research strategy is based on the characterization of the whole set of genes displaying non-additive expression to understand if gene regulation is related to functional or structural properties of the corresponding proteins. This question was addressed to some extent in synthetic allotetraploids of Arabidopsis suecica, at the transcript level using microarrays to identify the genes displaying expression divergence from the mid-parent value [11]. However, since mRNA abundances and protein amounts are poorly related [19], it is difficult to infer over- or under-representation of some protein functions and/or metabolic pathways from transcriptomic data alone. Thus, proteomic approaches constitute an alternative to determine if some functional categories or metabolic networks are over- or under-represented, or if the subcellular localization of the proteins is modified in an allopolyploid context.
In a previous work, we applied comparative proteomics to newly synthesized oilseed rape: Brassica napus allotetraploids were synthesized de novo from inter-specific hybridization between B. oleracea and B. rapa, followed by colchicine-induced chromosome doubling [14]. The additivity hypothesis (predicting a mid-parent proteome for the amphiploids) was tested in two distinct organs, the stem and root (see [14] for details). Most spots (519 in stem and 583 in root) displayed a mid-parent value in synthetic B. napus ("additive" polypeptides), while numerous others (335 in stem, 205 in root) displayed non-additive amounts [14]. Only a subset (~150) of these non-additive polypeptides were identified by mass spectrometry and submitted to in silico analyses [14]. In the present study, it is the whole set of non-additive proteins in synthetic B. napus (335 in the stem and 205 in the root, that is to say almost four hundreds supplementary "non-additive" polypeptides) that has been identified by mass spectrometry, as well as an additional two hundred "additive" proteins per organ that constitute the "control" group. Only such numerous data could allow an accurate and relevant analysis of the functional properties of the proteins in the synthetic allopolyploids. Thus, we report the thorough in silico analysis of protein functions, isoforms, complex subunits and subcellular localizations of the additive and non-additive proteins in synthetic amphiploids of B. napus.
Results
Comparative proteomics was applied to the stem and the root of synthetic B. napus and its diploid progenitors B. rapa and B. oleracea. The whole set of spots displaying non-additivity (i.e. spots whose abundance differed from the mid-parent value in the amphiploids) were excised (335 in stem, 205 in root, Table 1), as well as a subset of "additive" polypeptides randomly cut out (200 in stem, 200 in root). MALDI-TOF or nanoHPLC-MS/MS mass spectrometry allowed the identification of ~70% of the excised spots in both organs (see also complete protein identification results in Additional file 1).
Table 1.
Protein identification in synthetic Brassica napus.
| Stem | Root | |||||||
| Spots displaying additivity | Spots displaying non-additivity | Spots displaying additivity | Spots displaying non-additivity | |||||
| Number of picked spots | 200 | 335 | 200 | 205 | ||||
| Number of identified spots | 143 | 71.5% | 234 | 69.9% | 140 | 70.0% | 126 | 61.5% |
Spots were excised from two-dimensional electrophoresis gels and were identified by mass spectrometry (MALDI-TOF or nanoHPLC-MS/MS).
Protein isoforms can be differentially regulated
Protein identification from two-dimensional electrophoresis gels usually allows the detection of several isoforms [20,21]. Protein isoforms may be i- the results of post-translational modifications (PTMs) of the same gene product, ii- gene products of paralogous genes, iii- gene products of homeologous genes (i.e. orthologous genes from the two distinct parental species merged in a common nucleus via hybridization) and iv- a combination of these different origins. Since the expression of isoforms may be controlled by partially shared or conserved regulatory pathways, it can be hypothesized that they should display an identical regulation within the same organ (for example, a proportional increase in their quantity). This hypothesis was tested in synthetic Brassica napus: the identification of spots (displaying additivity and non-additivity) allowed the detection of groups of isoforms in both organs: 104 groups of isoforms in the stem and 82 in the root (Table 2). Among these groups, 42 to 48% comprised spots that all displayed an identical regulation within one organ (all isoforms up-regulated, or all isoforms down-regulated in the synthetic amphiploids compared to the mid parent value), in accordance with the above hypothesis. Interestingly, 15 to 24% of the groups included isoforms with "opposite" regulation patterns, resulting in partial or complete compensation: for example, in the stem, spots #1352 and #1353 were respectively B. oleracea- and B. rapa-specific and were both identified as putative fructokinases (Figure 1). These proteins were likely encoded by homeologous genes, and both displayed non-additive patterns in synthetic Brassica napus: spot #1352 was up-regulated compared to the mid parent value, and such an increase was partially balanced by the down-regulation of spot #1353. The differential regulation of homeologous gene products in allopolyploids appeared as a relatively frequent mechanism, since 7 out of 14 of the putative pairs of homeologous proteins displayed contrasting patterns of regulation.
Table 2.
Regulation of protein isoforms in synthetic Brassica napus.
| Stem | Root | |||
| Group of isoforms showing identical patterns of regulation | 44 | 42.3% | 39 | 47.6% |
| Group of isoforms showing opposite patterns of regulation | 25 | 24.0% | 12 | 14.6% |
| Group of isoforms showing different patterns of regulation | 35 | 33.7% | 31 | 37.8% |
| Total | 104 | 82 | ||
For each organ, protein identification data were used to assess isoforms. Groups of isoforms that comprised spots sharing the same pattern of regulation (i.e. all spots up-regulated compared to the mid parent value, all spots down-regulated or all spots displaying additivity) were designated as "Group of isoforms showing identical pattern of regulation". Groups of isoforms that encompassed up-regulated and down-regulated spots were designated as "Group of isoforms showing opposite patterns of regulation". Groups of isoforms that included additive and non-additive spots (amongst which non-additive spots shared identical patterns of regulation, i.e. up-regulation or down-regulation) were designated as "Group of isoforms showing different patterns of regulation".
Figure 1.
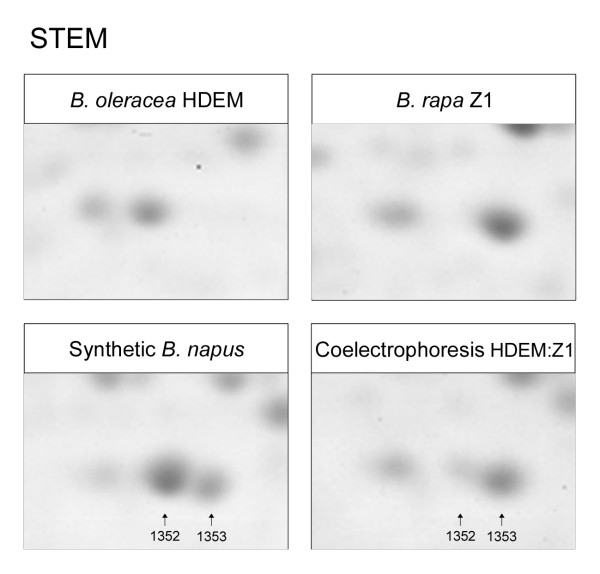
Opposite patterns of regulation between probable homeoallelic gene products. 2-DE gels of B. oleracea HDEM, B. rapa Z1, and synthetic B. napus stem proteomes. A coelectrophoresis of HDEM:Z1 stem extracts was added (mid-parent pattern). Spots #1352 and #1353 were HDEM- and Z1-specific respectively, and were both identified as putative fructokinases (At1g06020). These probable homeoallelic gene products displayed ''opposite'' patterns of regulation in synthetic Brassica napus in comparison to the mid-parent pattern: spot #1352 was up-regulated while spot #1353 was down-regulated.
One of the great advantages of comparative proteomics is the possibility to observe post-translational modifications (PTMs) of the same gene product. Although it is not obvious to demonstrate formally whether multiple isoforms correspond to PTMs or to products of homologous/homeologous genes, proteins with post-translational modifications such as phosphorylation usually appear on the 2-DE gels as horizontal rows of spots, resulting in characteristic "trains of spots". Figure 2 shows a train of four spots (#370, #374, #382 and #383) for which analysis by mass spectrometry led to the identification of one single accession, namely an Hsp70-like protein. Moreover, previous studies have described post-translational modifications of HSP 70-related proteins [22,23] so that the four detected spots most likely corresponded to different PTMs of HSP 70. Quantitative proteomic analysis revealed that spot #374 was additive in the synthetic B. napus, while isoform #370 was less abundant in amphiploids than expected under the additivity hypothesis whereas spots #382 and #383 were more abundant. Other examples of trains of spots with contrasting patterns of regulation were detected (26/44 for both organs), suggesting that the differential regulation of PTMs was not a rare event in the synthetic amphiploids of B. napus.
Figure 2.
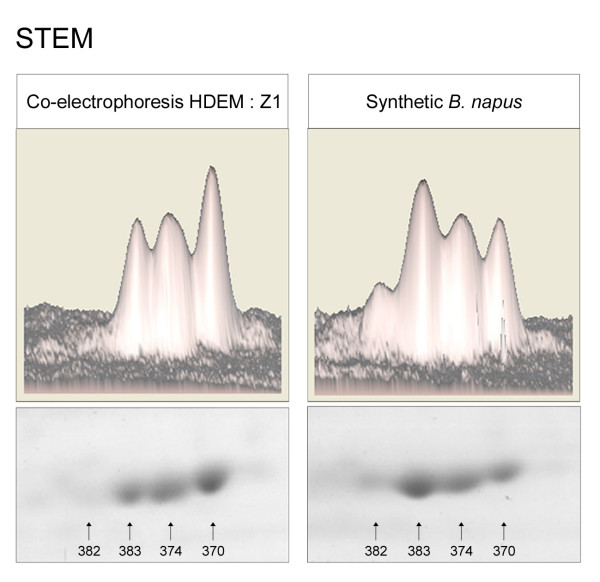
Opposite patterns of regulation between probable post-translationally modified isoforms. Two-dimensional and three-dimensional view of 2-DE stem gels. Spots #382, #383, #374 and #370 were identified by mass spectrometry as isoforms of heat shock protein 70-7 (At5g49910) and were probable post-translational modifications of the same gene product. Spot #374 displayed additivity in synthetic B. napus, while spot #370 was down-regulated and spots #382 and #383 were up-regulated compared to the mid parent value.
In order to know whether the distribution of the isoforms was similar between polypeptides displaying additivity and non-additivity, we counted the number of isoforms (Table 3). In both organs, significantly more isoforms were found among non-additive spots than among spots displaying additivity (χ2 tests, P-values = 3.76e-6 and 1.42e-10 in the stem and the root respectively, both inferior to the p-value = 0.05 limit). Such a difference might reflect the experimental bias due to the differential sampling of additive and non-additive spots. Since we analyzed by mass spectrometry all the spots with non-additive values, we detected all the groups within which at least two isoforms displayed non-additive patterns. On the other hand, we identified only a random subset of the additive spots (two hundred spots per organ while we found 519 and 583 additive spots in the stem and root respectively), so we may have underestimated the number of groups of isoforms within the additive group.
Table 3.
Protein isoforms amongst spots displaying additivity and non-additivity in Brassica napus.
| Stem | Root | |||||||
| Spots displaying additivity | Spots displaying non-additivity | Spots displaying additivity | Spots displaying non-additivity | |||||
| Number of identified spots | 143 | 234 | 140 | 126 | ||||
| Number of protein isoforms | 37 | 25.9% | 117 | 50.0% | 46 | 32.9% | 91 | 72.2% |
Significantly more isoforms were found amongst spots displaying non-additivity than amongst spots displaying additivity (χ2 tests, P-values < 0.05, P-value = 3.76e-6 in the stem and P-value = 1.42e-10 in the root).
Complex subunits can be differentially regulated
Most metabolic pathways are driven by protein complexes. Thus, the stoichiometry of the different subunits might be a critical issue for cell processing, and the components of a protein complex are expected to display parallel variations of their respective amounts. These supposed co-regulation and coordinate expressions are known as "the balance hypothesis", and it has been suggested that both under-expression and over-expression (imbalance) of subunits shall be deleterious [24,25]. Hence, the "balance hypothesis" was tested in the synthetic amphiploids of Brassica napus: protein complexes with at least two identified distinct subunits were studied. Eleven and nine complexes were found in the stem and root respectively, and the pattern of regulation of their components was investigated (Table 4). For example, analysis of the stem proteome led to the identification of the alpha, beta, delta and gamma subunits of the chloroplastic ATP synthase, and all of these four subunits displayed non-additive up-regulation in B. napus synthetic amphiploids compared to the mid parent value. On the contrary, two subunits of the ribosomal complex, the 40S ribosomal protein SA (RPSaA) and the 60S acidic ribosomal protein P0 (RPP0A) [26], were found to be differentially expressed in the root, but with opposite patterns of regulation: two isoforms of RPSaA subunit were down-regulated while RPP0A was up-regulated in the amphiploids (Figure 3). Finally, only 5/11 complexes in stem and 2/9 in root encompassed subunits with identical patterns of regulation, demonstrating that the different subcomponents of a complex can be differentially regulated in neo-synthetized B. napus. These results suggest that the balance hypothesis might not be a general rule for the regulation of different complex subunits in synthetic allopolyploids.
Table 4.
Regulation of complex subunits in synthetic Brassica napus.
| Complexes with at least two identified subunits | Stem | root |
| Complexes with subunits showing identical regulation patterns | 5 | 2 |
| Complexes with subunits showing opposite regulation patterns | 1 | 3 |
| Complexes with subunits showing differing regulation patterns | 5 | 4 |
| Total | 11 | 9 |
For each organ, complexes with at least two distinct subunits identified were detected. Those complexes were differentiated on the basis of the regulation patterns of the subunits that could either display an identical pattern of regulation (all subunits up-regulated compared to the mid parent value, or all subunits down-regulated), or show opposite regulation patterns (for example, one subunit up-regulated, the other one down-regulated). Complexes comprising subunits with non-additive patterns and subunits with additive patterns were designated as "Complexes with subunits showing differing regulation patterns".
Figure 3.
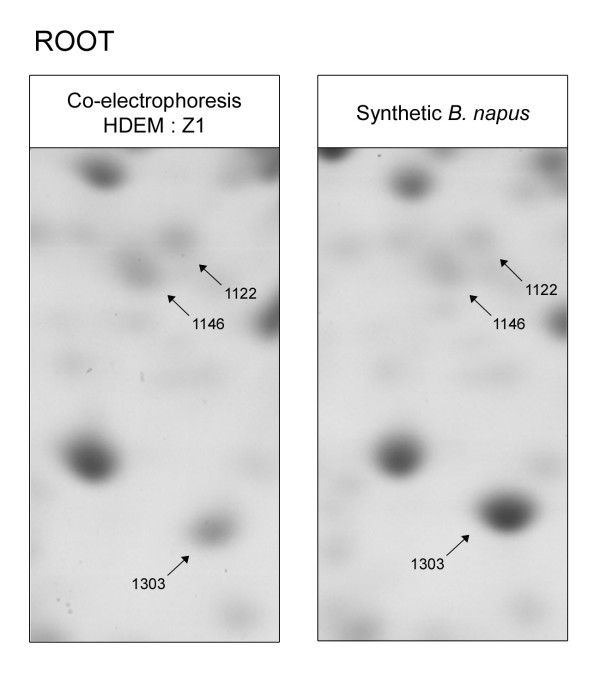
Opposite patterns of regulation between RPSaA and RPP0A, small and large ribosome subunits. Spots #1122 and #1146 were identified by mass spectrometry as isoforms of 40S ribosomal protein SA RPSaA (At1g72370). Both spots were down-regulated in synthetic B. napus compared to the mid parent value. Spot #1303 was identified as a 60S acidic ribosomal protein P0 RPP0A (At2g40010) and was up-regulated in the amphiploids. RPSaA and RPP0A were respectively small and large ribosome subunits [26].
No functional categories are under- or over-represented among proteins displaying non-additivity in synthetic B. napus
One of the major goals of protein identification of both additive and non-additive spots was to find whether some functions or metabolic pathways were preferentially affected by allopolyploidization. Thus, the functional categorization of the identified polypeptides was carried out using the MIPS FunCat database available on line [27]. Since the proportion of isoforms was significantly imbalanced between additive and non-additive spots, we did not consider the isoforms individually but we counted each group of isoforms as a single protein. Figure 4 shows the distribution of additive and non-additive spots into functional categories. In both organs, the identified proteins were mainly involved in metabolism (26% to 37%) and energy (14% to 21%), which was congruent with previous proteomic data [21,28]. Protein fate, transport, defense and biogenesis of cellular components were other well represented functional categories (5% to 10%), while an important proportion of the identified polypeptides were not assigned to any known function (30% to 42%). No functional category was found over- or under-represented for spots displaying non-additivity compared to "additive" spots (χ2 tests, p-values > 0.05), suggesting that differential regulation of gene products in synthetic Brassica napus was not related to the function of the proteins.
Figure 4.
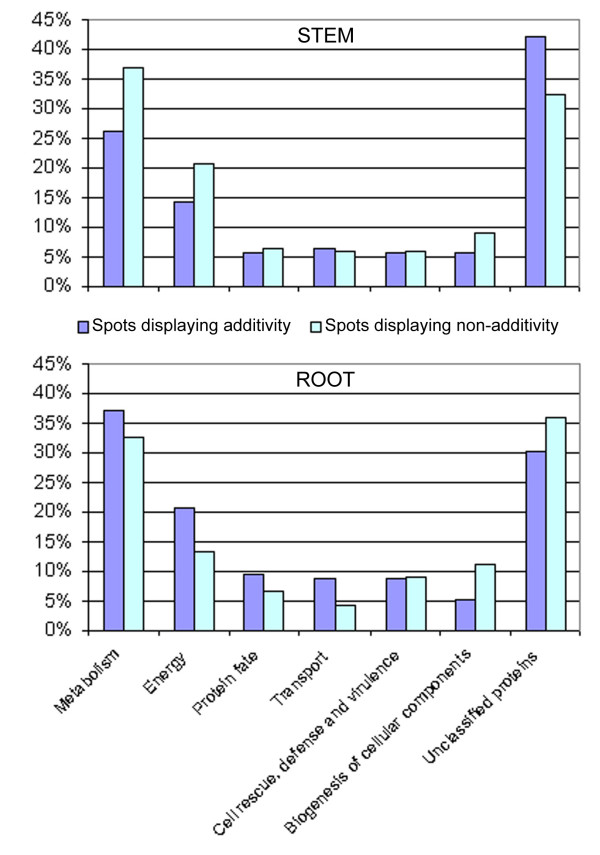
Functional categorization of the proteins identified in synthetic Brassica napus. Each group of isoforms was counted once: in the stem, among the 143 proteins displaying additivity in the amphiploids, 126 were non-redundant and were taken into account. For non-additive spots, 155/234 non-additive spots were non-redundant and analyzed. In the root, 115/140 additive and 89/126 non-additive spots were non-redundant and were taken into account. Functional categorization was made using the MIPS FunCat database [27]. The "transport" category covers cellular transport, transport facilitation and transport routes.
In addition, we used the KEGG PATHWAY database [29] to identify the proteins involved in the same metabolic pathways (Table 5). No pathway was particularly altered by differential gene expression, since no proteins involved in the same pathway were found all up-regulated, or down-regulated in the synthetic amphiploids compared to the mid parent value. Moreover, the proteins involved in the same pathway could display different patterns of regulation: for example, most of the glycolytic enzymes were identified in stem proteome (Figure 5). Some of these proteins were up-regulated in the amphiploids (like the phosphoglucoisomerase), others were down-regulated (enolase) or displayed an additive pattern (phosphoglyceromutase). Taken together, these results indicated that neither functional category nor metabolic pathway were particularly targeted by differential regulation of gene expression in the amphiploids.
Table 5.
The main metabolic pathways identified in stem and root proteomes.
| Stem | Spots displaying additivity | Spots displaying non-additivity | ||
| Glycolysis/Gluconeogenesis | 6 | 4.8% | 11 | 7.1% |
| Citrate cycle (TCA cycle) | 7 | 5.6% | 6 | 3.9% |
| Pyruvate metabolism | 5 | 4.0% | 8 | 5.2% |
| Photosynthesis | 5 | 4.0% | 9 | 5.8% |
| Carbon fixation | 5 | 4.0% | 15 | 9.7% |
| Proteasome | 4 | 3.2% | 8 | 5.2% |
| Number of non-redundant proteins | 126 | 155 | ||
| Root | Spots displaying additivity | Spots displaying non-additivity | ||
| Glycolysis/Gluconeogenesis | 9 | 7.8% | 3 | 3.4% |
| Citrate cycle (TCA cycle) | 9 | 7.8% | 6 | 6.7% |
| Pyruvate metabolism | 8 | 7.0% | 4 | 4.5% |
| Carbon fixation | 7 | 6.1% | 5 | 5.6% |
| Number of non-redundant proteins | 115 | 89 | ||
For both organs, the main molecular networks were identified using the KEGG PATHWAY database [29]. No bias was found between spots fitting additivity and spots displaying non-additivity in synthetic B. napus (χ2 tests. P-value > 0.05).
Figure 5.
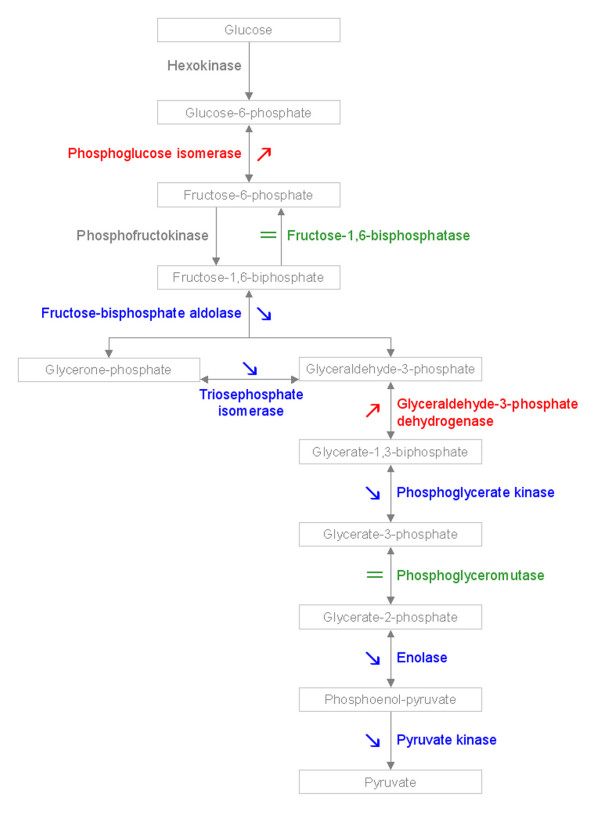
Patterns of regulation of glycolytic enzymes in synthetic Brassica napus. Enzymes up-regulated in synthetic B. napus compared to the mid parent value are colored in red, enzymes down-regulated are marked by blue scripts, and enzymes displaying an additive pattern are green in color. The hexokinase and the phosphofructokinase were not identified. The fructose-biphosphate aldolase was globally down-regulated, since four isoforms were found: one being up-regulated, the three others being down-regulated. For the triosephosphate isomerase, two isoforms were found, both of them being down-regulated. For the other enzymes, only one isoform was identified.
Proteins displaying non-additivity in synthetic B. napus are not from a peculiar cellular localization
The subcellular localization of the proteins was assessed using the Gene Ontology GO annotations [30]. As for the functional categorization of the proteins, each group of isoforms was taken into account only once in order to correct for the bias between additive and non-additive spots. Figure 6 shows the distribution of the identified proteins within cellular compartments: in both organs, most of the polypeptides were located in the organelles (chloroplast and mitochondria), in accordance with the main functional categories previously found, namely Energy and Metabolism. The fact that ~10% of the root proteins were located in the "chloroplast" actually indicate that these proteins were located in the plastids, and particularly in the amyloplasts, which are very abundant in root tissues [31]. Other cellular localizations, such as cytosol, membrane and nucleus, were well represented in both organs, indicating that comparative proteomics allows the analysis of all the compartments of the cell. The comparison between non-additive and additive spots showed no specific subcellular localization (χ2 tests, p-values > 0.05), which suggests that the regulation of gene products in the amphiploids was not related to subcellular localization.
Figure 6.
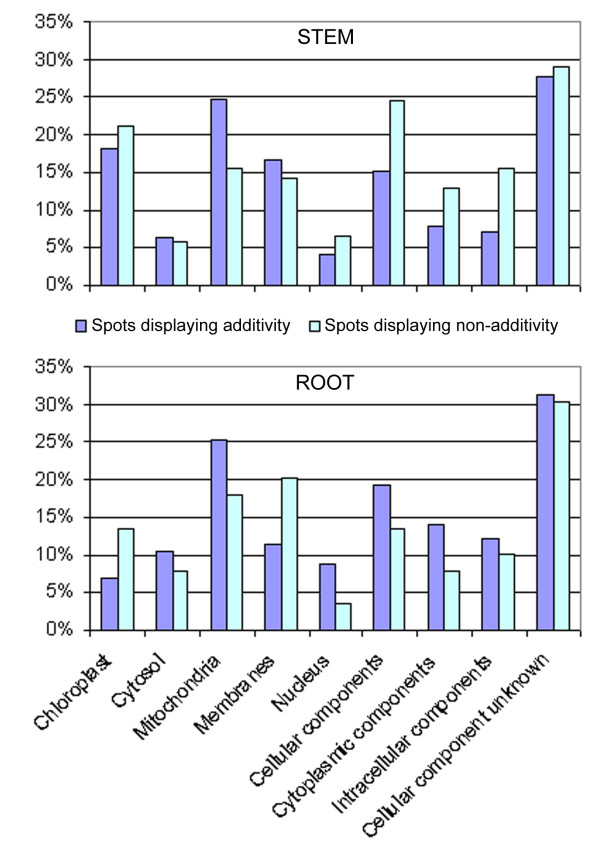
Cellular localization of the proteins identified in synthetic Brassica napus. Each group of isoforms was counted once: in the stem, 126/143 additive and 155/234 non-additive spots were non-redundant and were taken into account. In the root, 115/140 additive and 89/126 non-additive spots were non-redundant and were taken into account. The cellular localization of the proteins was assessed using the Gene Ontology GO annotations [30].
Discussion
The differential regulation of isoforms and complex subunits modifies the equilibrium between functionally related proteins in synthetic Brassica napus
Protein identification in synthetic amphiploids of B. napus allowed the discovery of several isoforms that could display different patterns of regulation. In particular, probable homeoallelic products were found with "opposite" regulation (up- and down-regulation in the synthetic allotetraploids compared to the mid parent value), suggesting that differential expression of homeologous genes could occur. Unequal contributions of homeoalleles to the transcriptome have already been evidenced in synthetic allotetraploids of Gossypium [12,32]. Here, we have shown that such a phenomenon was detectable at the proteomic level and was not a rare event. Moreover, our proteomic data suggest that the post-translational modifications of the same gene product could also be differentially regulated in the amphiploids. Differential regulation of PTMs has been described in a few proteomic analyses [33], but no data was available in the context of allopolyploidy. PTMs play a key role in the cell as the same gene product may have different activities, modified turn-overs, distinct subcellular locations, or altered interactions with proteins or nucleic acids depending on its PTMs. Thus, variation of amounts among protein isoforms (PTMs, but also homeoalleles or paralogous genes) may establish a novel metabolic equilibrium in synthetic Brassica napus in contrast to its diploid progenitors.
Complex subunits are usually supposed to present co-ordinate expressions [24,25]. Such a statement was supported by some experimental data showing that genes encoding subcomponents of a complex displayed the same pattern of regulation of their expressions [34,35]. In contradiction with this assumption, we have shown that the different subunits of a protein complex could be differentially regulated in newly synthesized B. napus. This result may reflect the instability of protein regulation in the very first step of allopolyploidization, and it will be of particular interest to investigate the evolution of such imbalance in the further generations of allotetraploids. Moreover, although the synthetic B. napus plants we analyzed were viable and morphologically "normal", neither phenotypic analyses nor relative fitness studies were undertaken. Thus, the imbalanced levels of isoforms and subunit complexes may have an impact on the fitness and/or the phenotype of the neo-allopolyploids that remains to be quantified. One can also suppose that, due to genetic and proteomic redundancies, expression constraints are relaxed within an amphiploid, allowing previously constrained genes to be differentially regulated and to evolve in a different way.
Differential regulation of gene products is not related to functional properties of the proteins
During the last few years, several synthetic allopolyploids have been investigated at the transcriptional level using various approaches (cDNA-SSCP in synthetic Gossypium allotetraploids [12], AFLP- cDNA in synthetic wheat allopolyploids [15], microarray in synthetic Senecio allopolyploids [13] and synthetic Gossypium allotetraploids [32], etc.). Non-additive transcription in hybrids and allopolyploids is now well documented, yet the genes concerned by non-additive expression are not well characterized. Recently, a transcriptional analysis of Arabidopsis allotetraploids showed that genes involved in "hormonal regulation" and in "cell defense and aging" were more susceptible to expression changes [11]. In addition, the ethylene biosynthesis pathway was found globally repressed in two distinct lineages of synthetic A. suecica [11]. In this report, we have described a thorough in silico functional analysis of proteins in synthetic B. napus, and we have shown that neither specific functional category nor metabolic pathway were over- or under-represented among non-additive proteins. This discrepancy between Arabidopsis transcriptional results and oilseed rape proteomic data may be explained by the fact that mRNA abundance and protein amounts are poorly related [19]) or by the reference used (we compared the functional categorizations of non-additive proteins to additive ones, while Wang et al. compared the distribution among functional categories of non-additive genes to the whole set of 26,000 annotated genes in Arabidopsis [11]). Moreover, differential regulation of gene expression may vary from one biological model to an other (Arabidopsis vs. Brassica).
Finally, this is the first in silico analysis of proteins in synthetic allotetraploids. Although in silico prediction is not a perfect process and may give erroneous information for some genes considered individually [36], bioinformatic tools are particularly relevant for genome-wide analysis and global consideration of the data. We have shown that non-additive and additive polypeptides could not be differentiated on the basis of their function nor subcellular localization, indicating that the regulation of gene products following allopolyploidization is not related to any functional feature of the proteins.
Allopolyploidy: a genomic shock but not a proteomic shock?
Polyploidy is often described as a genomic shock [37,38], that may induce stress and defense responses. Our study showed that, in synthetic B. napus, the "cell rescue, defense and virulence" category was unchanged in the stem and root. In addition, Wang et al. showed that the "defense" class was under-represented when analyzing differential gene transcription in leaves and flower buds of synthetic Arabidopsis allotetraploids [11]. These results suggest that neither stress nor defense responses were particularly enhanced following a recent polyploidy event for these two Brassicaceae species. Moreover, although stem and root proteomes were extensively modified in synthetic B. napus, the patterns were reproducible in independent amphiploid lineages [14] and our data indicated that, globally, the distribution of the proteins into metabolic pathways, functional categories and subcellular localizations was not disturbed. Thus, at the protein level, there is no evidence for a chaos or a large disorder following the merging of two genomes. Instead, a novel order establishes quickly and may evolve in further generations of synthetic B. napus. Hence, allopolyploidy can be seen as a mechanism making new from old: new expression patterns from old genomes, and perhaps this feature explains in part why polyploidy is such an undeniable evolutionary success.
Differential gene expression: what are the underlying mechanisms?
The thorough in silico analysis of the non-additive proteins in synthetic B. napus indicated that the differential regulation of gene products was not related to functional properties of the proteins. The molecular mechanisms however remain to be investigated, such as the possible implication of transposable elements: more and more data suggest that transposable elements have played a key role in polyploid evolution such as Gossypium or Arabidopsis [9,39], and that their transcriptional activation could alter the expression of adjacent genes (like in wheat allopolyploids [40]). Epigenetic mechanisms (changes in chromatin structure, histone modification, DNA methylation, etc.) are described in synthetic allopolyploids [41,42] and may be responsible for the modification of gene expression in polyploids [43,44]. Furthermore, RNA interference, action of siRNAs and miRNAs and their implication in gene regulation, are thus good candidates to explain nonadditive gene expression [45]. Finally, allopolyploidy not only induces the merger of homeologous genomes in a common nucleus, but also the juxtaposition of divergent regulatory networks. The interaction and the coordination of such homeologous networks in a newly synthesized allopolyploid may also explain the genome-wide modification of gene expression, as well as the maintenance at a viable level, in successful individual plants, of the equilibrium between the different functions, metabolic pathways and cellular localizations.
Conclusion
The complete set of proteins displaying non-additivity and a random sample of additive polypeptides were identified by mass spectrometry in synthetic Brassica napus. Functionally related proteins, such as isoforms and complex subunits, were detected and some of them displayed different or opposite patterns of regulation. No functional category, no metabolic pathway and no subcellular localization was found over- or under-represented for non-additive polypeptides. These results suggested that the differential regulation of gene products was not related to functional properties of the proteins and that the mechanisms responsible for the regulation of gene expression in synthetic amphiploids (transposon activation ?, epigenetic processes?) were not initiated according to any protein characteristic. To better understand the success of polyploidy, additional work based on transcriptional analysis and study of the post-translational modifications of the non-additive proteins will be necessary. Only such an integrative study would help in understanding the molecular and biochemical mechanisms responsible for the regulation of gene expression which occurs in a polyploid organism.
Methods
Mass spectrometry identification of additive and non-additive proteins
In a previous report, we applied comparative proteomics to synthetic Brassica napus (four independent lineages) and its diploid progenitors B. rapa and B. oleracea (two to five plants analyzed per genotype) [14]. Our proteomic analysis showed that several spots (519 in stem and 583 in root) displayed a mid-parent value in synthetic Brassica napus compared to its diploid progenitors while many others (335 in stem, 205 in root) displayed non-additive amounts [14]. About 150 of these non-additive polypeptides were identified by mass spectrometry and submitted to in silico analyses [14]. However, the subset of non-additive polypeptides analyzed was too small to obtain accurate and relevant data on the functional categorization and subcellular localization of the non-additive proteins [14]. Here, the remaining non-additive spots (~260 in stem and ~130 in root) were excised from 2-DE gels and submitted to mass spectrometry, so that the whole set of non-additive proteins in synthetic B. napus was identified.
In our previous study, no accurate "control group" was available (no additive spot was identified), so that we used reference proteomic maps of other organisms (Pisum sativum, Medicago truncatula) [14]. Here, two hundred spots displaying additivity were also cut out for both organs (random excision among the 519 and 583 spots found additive in stem and root [14]). These additive polypeptides were used as controls for subsequent in silico analyses. Mass spectrometry analyses were conducted as described in [14].
Assessing protein isoforms
Distinct spots were considered as isoforms when they met the following criteria: 1 – the corresponding identified protein sequences presented more than 70% identity all along the sequences; 2 – the corresponding proteins had the same function (see below) 3 – the polypeptides were assessed in silico to the same cellular compartment (see cellular localization below) and 4 – the spots displayed similar molecular weight on 2-DE gels (to discard degradation or catabolic products).
In silico analyses
Functional categorization and cellular localization of the proteins were conducted using the MIPS FunCat database (Version 2.0) available on line [27] and the TAIR's Gene Ontology (GO) annotations (release of February 2006) [46,47] as described in [14]. For both organs, the main molecular networks were identified using the KEGG PATHWAY database (release of February 2006) [29]. χ2 tests were carried out to test statistical differences between proteins fitting additivity or displaying non-additivity.
Abbreviations
2-DE: two-dimensional electrophoresis ; PTM: post-translational modification
Authors' contributions
WA carried out comparative proteomics, analyzed the mass spectra, made in silico analyses and drafted the manuscript. KA and PB participated in the design of the study, the interpretation of the data and helped to draft the manuscript. TB, MD and BV carried out nanoHPLC-MS/MS analyses; CM carried out MALDI-TOF analyses. HT conceived and designed the study, and drafted the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Protein identification in synthetic Brassica napus. Patterns: "additive": spot displaying a mid-parent value in the amphiploid compared to its diploid progenitors ; "positive/negative over-dominance": spot displaying a higher/lesser quantification value in the amphiploid compared to the higher/lesser parent ; "HDEM or Z1 dominance": amphiploid spot abundance equaled either HDEM or Z1 value ; "up-regulation" or "down-regulation": spot amount superior or inferior to the mid-parent value ; "Intermediary" pattern: non-additive spot with an abundance ranging between mid-parent and parental values; "progressive": spot abundance increased or decreased progressively with successive amphiploid generations.
Acknowledgments
Acknowledgements
We thank our colleagues (the GEAR and GQMP teams, the UMR de Génétique Végétale, Gif-sur-Yvette) for their help, technical advice and knowledge. We are very grateful to Anne-Marie Chèvre, Frédérique Eber and Jean-Claude Letanneur (UMR ENSAR-INRA, Le Rheu, France) for creating and providing the biological material. We thank Catherine Damerval for careful reading of the manuscript, and the anonymous reviewers for helpful suggestions.
W. Albertin was supported by a thesis grant from the French Ministère de la Jeunesse, de l'Education Nationale et de la Recherche. The Plate-forme de Protéomique du Moulon and its equipment were supported by grants from the Région Ile-de-France, IFR 87, Génoplante, the INRA and the CNRS. The Plate-forme de Protéomique de Versailles and its equipment were supported by grants from the Région Ile-de-France and the INRA. Polyploid research was supported by the grant # ANR-05-BDIV-015-03 from the Agence Nationale de la Recherche (ANR) Biodiversity programme.
Contributor Information
Warren Albertin, Email: albertin@moulon.inra.fr.
Karine Alix, Email: alix@moulon.inra.fr.
Thierry Balliau, Email: balliau@moulon.inra.fr.
Philippe Brabant, Email: brabant@moulon.inra.fr.
Marlène Davanture, Email: davanture@moulon.inra.fr.
Christian Malosse, Email: malosse@dcmr.polytechnique.fr.
Benoît Valot, Email: valot@moulon.inra.fr.
Hervé Thiellement, Email: thiellement@moulon.inra.fr.
References
- Wendel JF. Genome evolution in polyploids. Plant Mol Biol. 2000;42:225–249. doi: 10.1023/A:1006392424384. [DOI] [PubMed] [Google Scholar]
- Adams KL, Wendel JF. Polyploidy and genome evolution in plants. Curr Opin Plant Biol. 2005;8:1–7. doi: 10.1016/j.pbi.2005.01.001. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Moore RC, Purugganan MD. The evolutionary dynamics of plant duplicate genes. Curr Opin Plant Biol. 2005;8:122–128. doi: 10.1016/j.pbi.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Paterson AH. Polyploidy, evolutionary opportunity, and crop adaptation. Genetica. 2005;123:191–196. doi: 10.1007/s10709-003-2742-0. [DOI] [PubMed] [Google Scholar]
- Otto SP, Whitton J. Polyploid incidence and evolution. Annu Rev Genet. 2000;34:401–437. doi: 10.1146/annurev.genet.34.1.401. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Soltis PS, Tate JA. Advances in the study of polyploidy since Plant speciation. New Phytologist. 2003;161:173–191. doi: 10.1046/j.1469-8137.2003.00948.x. [DOI] [Google Scholar]
- Albertin W, Brabant P, Catrice O, Eber F, Jenczewski E, Chevre AM, Thiellement H. Autopolyploidy in cabbage (Brassica oleracea L.) does not alter significantly the proteomes of green tissues. Proteomics. 2005;5:2131–2139. doi: 10.1002/pmic.200401092. [DOI] [PubMed] [Google Scholar]
- Madlung A, Tyagi AP, Watson B, Jiang H, Kagochi T, Doerge RW, Martienssen R, Comai L. Genomic changes in synthetic Arabidopsis polyploids. The Plant Journal. 2005;41:221–230. doi: 10.1111/j.1365-313X.2004.02297.x. [DOI] [PubMed] [Google Scholar]
- Comai L, Tyagi AP, Winter K, Holmes-Davis R, Reynolds SH, Stevens Y, Byers B. Phenotypic instability and rapid gene silencing in newly formed arabidopsis allotetraploids. Plant Cell. 2000;12:1551–1568. doi: 10.1105/tpc.12.9.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tian L, Lee HS, Wei NE, Jiang H, Watson B, Madlung A, Osborn TC, Doerge RW, Comai L, Chen ZJ. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics. 2006;172:507–517. doi: 10.1534/genetics.105.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams KL, Cronn R, Percifield R, Wendel JF. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc Natl Acad Sci U S A. 2003;100:4649–4654. doi: 10.1073/pnas.0630618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegarty MJ, Jones JM, Wilson ID, Barker GL, Coghill JA, Sanchez-Baracaldo P, Liu G, Buggs RJ, Abbott RJ, Edwards KJ, Hiscock SJ. Development of anonymous cDNA microarrays to study changes to the Senecio floral transcriptome during hybrid speciation. Mol Ecol. 2005;14:2493–2510. doi: 10.1111/j.1365-294x.2005.02608.x. [DOI] [PubMed] [Google Scholar]
- Albertin W, Balliau T, Brabant P, Chèvre AM, Eber F, Malosse C, Thiellement H. Numerous and rapid non-stochastic modifications of gene products in newly synthesized Brassica napus allotetraploids. Genetics. 2006;173:1101–1113. doi: 10.1534/genetics.106.057554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Friebe BR, Gill BS, Zhou JM. Allopolyploidy alters gene expression in the highly stable hexaploid wheat. Plant Mol Biol. 2003;52:401–414. doi: 10.1023/A:1023965400532. [DOI] [PubMed] [Google Scholar]
- Adams KL, Percifield R, Wendel JF. Organ-specific silencing of duplicated genes in a newly synthesized cotton allotetraploid. Genetics. 2004;168:2217–2226. doi: 10.1534/genetics.104.033522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tian L, Madlung A, Lee HS, Chen M, Lee JJ, Watson B, Kagochi T, Comai L, Chen ZJ. Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics. 2004;167:1961–1973. doi: 10.1534/genetics.104.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlung A, Comai L. The effect of stress on genome regulation and structure. Ann Bot (Lond) 2004;94:481–495. doi: 10.1093/aob/mch172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechin V, Balliau T, Chateau-Joubert S, Davanture M, Langella O, Negroni L, Prioul JL, Thevenot C, Zivy M, Damerval C. A two-dimensional proteome map of maize endosperm. Phytochemistry. 2004;65:1609–1618. doi: 10.1016/j.phytochem.2004.04.035. [DOI] [PubMed] [Google Scholar]
- Schiltz S, Gallardo K, Huart M, Negroni L, Sommerer N, Burstin J. Proteome reference maps of vegetative tissues in pea. An investigation of nitrogen mobilization from leaves during seed filling. Plant Physiol. 2004;135:2241–2260. doi: 10.1104/pp.104.041947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A, Matusiewicz N, Stochaj U. Heat-induced nuclear accumulation of hsc70s is regulated by phosphorylation and inhibited in confluent cells. Faseb J. 2001;15:1478–1480. doi: 10.1096/fj.00-0680fje. [DOI] [PubMed] [Google Scholar]
- Kumar S, Hedges SB. A molecular timescale for vertebrate evolution. Nature. 1998;392:917–920. doi: 10.1038/31927. [DOI] [PubMed] [Google Scholar]
- Papp B, Pal C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424:194–197. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- Veitia RA. Gene dosage balance: deletions, duplications and dominance. Trends Genet. 2005;21:33–35. doi: 10.1016/j.tig.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Nakao A, Yoshihama M, Kenmochi N. RPG: the Ribosomal Protein Gene database. Nucleic Acids Res. 2004;32:D168–70. doi: 10.1093/nar/gkh004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The MIPS FunCat database http://mips.gsf.de/proj/funcatDB/
- Mathesius U, Keijzers G, Natera SH, Weinman JJ, Djordjevic MA, Rolfe BG. Establishment of a root proteome reference map for the model legume Medicago truncatula using the expressed sequence tag database for peptide mass fingerprinting. Proteomics. 2001;1:1424–1440. doi: 10.1002/1615-9861(200111)1:11<1424::AID-PROT1424>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- The KEGG PATHWAY database http://www.genome.jp/kegg/pathway.html
- The Gene Ontology GO annotations database http://www.arabidopsis.org/tools/bulk/go/
- Deng XW, Gruissem W. Constitutive transcription and regulation of gene expression in non-photosynthetic plastids of higher plants. Embo J. 1988;7:3301–3308. doi: 10.1002/j.1460-2075.1988.tb03200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udall JA, Swanson JM, Nettleton D, Percifield RJ, Wendel JF. A novel approach for characterizing expression levels of genes duplicated by polyploidy. Genetics. 2006;in press doi: 10.1534/genetics.106.058271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent D, Lapierre C, Pollet B, Cornic G, Negroni L, Zivy M. Water deficits affect caffeate O-methyltransferase, lignification, and related enzymes in maize leaves. A proteomic investigation. Plant Physiol. 2005;137:949–960. doi: 10.1104/pp.104.050815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos B, Meiklejohn CD, Hartl DL. Regulatory evolution across the protein interaction network. Nat Genet. 2004;36:1059–1060. doi: 10.1038/ng1427. [DOI] [PubMed] [Google Scholar]
- Marri L, Sparla F, Pupillo P, Trost P. Co-ordinated gene expression of photosynthetic glyceraldehyde-3-phosphate dehydrogenase, phosphoribulokinase, and CP12 in Arabidopsis thaliana. J Exp Bot. 2005;56:73–80. doi: 10.1093/jxb/eri020. [DOI] [PubMed] [Google Scholar]
- Hogenesch JB, Ching KA, Batalov S, Su AI, Walker JR, Zhou Y, Kay SA, Schultz PG, Cooke MP. A comparison of the Celera and Ensembl predicted gene sets reveals little overlap in novel genes. Cell. 2001;106:413–415. doi: 10.1016/S0092-8674(01)00467-6. [DOI] [PubMed] [Google Scholar]
- McClintock B. The significance of responses of the genome to challenge. Science. 1984;226:792–801. doi: 10.1126/science.15739260. [DOI] [PubMed] [Google Scholar]
- Comai L, Madlung A, Josefsson C, Tyagi A. Do the different parental 'heteromes' cause genomic shock in newly formed allopolyploids? Philos Trans R Soc Lond B Biol Sci. 2003;358:1149–1155. doi: 10.1098/rstb.2003.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006 doi: 10.1101/gr.5282906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashkush K, Feldman M, Levy AA. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat Genet. 2003;33:102–106. doi: 10.1038/ng1063. [DOI] [PubMed] [Google Scholar]
- Liu B, Vega JM, Feldman M. Rapid genomic changes in newly synthesized amphiploids of Triticum and Aegilops. II. Changes in low-copy coding DNA sequences. Genome. 1998;41:535–542. doi: 10.1139/gen-41-4-535. [DOI] [PubMed] [Google Scholar]
- Song K, Lu P, Tang K, Osborn TC. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proc Natl Acad Sci U S A. 1995;92:7719–7723. doi: 10.1073/pnas.92.17.7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Wang J, Tian L, Lee HS, Wang JJ, Chen M, Lee JJ, Josefsson C, Madlung A, Watson B, Lippman Z, Vaughn M, Pires JC, Colot V, Doerge RW, Martienssen RA, Comai L, Osborn TC. The development of an Arabidopsis model system for genome-wide analysis of polyploidy effects. Biol J Linn Soc Lond. 2004;82:689–700. doi: 10.1111/j.1095-8312.2004.00351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn TC, Chris Pires J, Birchler JA, Auger DL, Jeffery Chen Z, Lee HS, Comai L, Madlung A, Doerge RW, Colot V, Martienssen RA. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 2003;19:141–147. doi: 10.1016/S0168-9525(03)00015-5. [DOI] [PubMed] [Google Scholar]
- Matzke MA, Birchler JA. RNAi-mediated pathways in the nucleus. Nat Rev Genet. 2005;6:24–35. doi: 10.1038/nrg1500. [DOI] [PubMed] [Google Scholar]
- Berardini TZ, Mundodi S, Reiser L, Huala E, Garcia-Hernandez M, Zhang P, Mueller LA, Yoon J, Doyle A, Lander G, Moseyko N, Yoo D, Xu I, Zoeckler B, Montoya M, Miller N, Weems D, Rhee SY. Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol. 2004;135:745–755. doi: 10.1104/pp.104.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Protein identification in synthetic Brassica napus. Patterns: "additive": spot displaying a mid-parent value in the amphiploid compared to its diploid progenitors ; "positive/negative over-dominance": spot displaying a higher/lesser quantification value in the amphiploid compared to the higher/lesser parent ; "HDEM or Z1 dominance": amphiploid spot abundance equaled either HDEM or Z1 value ; "up-regulation" or "down-regulation": spot amount superior or inferior to the mid-parent value ; "Intermediary" pattern: non-additive spot with an abundance ranging between mid-parent and parental values; "progressive": spot abundance increased or decreased progressively with successive amphiploid generations.