

Abstract
Despite important recent insights into the short-term dynamics of HIV-1 infection, our understanding of the long-term pathogenesis of AIDS remains unclear. Using an approach that places rapid progressors, typical progressors, and nonprogressors on a single clinical spectrum of disease progression, we quantitate the previously reported relationship between viral load and survival time. We introduce the concept of viral constant, present evidence that this quantity is conserved across patients, and explore the immunopathological implications of this finding. We conclude with a quantitative approach for assessing the benefits of a given regime of antiviral therapy.
Keywords: prognosis, CD4+ cell count, CD4+ cell decline, antiviral therapy, statistical analysis
HIV-1 infection is marked by a progressive decline in the number of circulating CD4+ T helper cells, which over a period of years leads to death from immune failure and opportunistic infection (1, 2). Although its clinical course is known to vary considerably from patient to patient, with progression to AIDS taking anywhere from 2 to 10 or more years, the reason for this variability remains unclear.
In addressing this question, several studies have reported a link between fast progression and high viral load in the blood (3–8). However, because of the great range in viral load measurements between patients (9–14), these studies have tended to group patients based on whether their measurements are high, middling, or low (3–7). Still, that viral load and disease progression should be linked in the aggregate makes it natural to suspect that they are linked at the level of the individual, and that there is some underlying relationship between viral load and survival time that holds true for patients from across the clinical spectrum. We present evidence that such a relationship exists and explore its potential immunopathological and therapeutic implications.
MATERIALS AND METHODS
Patient data were drawn from a National Cancer Institute study that began in 1982 of 134 HIV-1 seropositive men (10, 15). Viral load, CD4+ cell count, and clinical and treatment status were recorded on average once a year for each patient. For the 50 patients who seroconverted during the study, dates of seroconversion were estimated as the midpoint between the dates of the last negative and first positive viral load measurements, resulting in a measurement error of at most ± approximately 1 year for 47 of the 50. The remaining 84 patients were seropositive at the start of the study, making precise estimation of seroconversion dates impossible; seroconversion dates for these patients instead were estimated from epidemiological data on the cities in which they lived (15).
Of the 47 patients whose seroconversion dates could be well estimated, 16 progressed to AIDS and died during the study. Survival times, as approximated by the time from seroconversion until death, ranged from 3.8 to 13.7 years, with a mean of 6.9 years, consistent with previous reports (Table 1) (2). Of these 16, five received some form of antiviral treatment (though not triple-drug therapy), but in no case did this significantly affect final disease outcome. In general, each patient’s viral load measurements varied less than 10-fold after the resolution of primary infection. In most patients it followed no significant trend, although in six it increased moderately over time, consistent with previous reports (Fig. 1) (9–11).
Table 1.
Survival time, average viral load, and viral constant
| Patient | ta, years | v̄, copies HIV-1 RNA/ml × 104 | Viral constant, viral years |
|---|---|---|---|
| 108† | 3.8 | 11.3 | 1,128 |
| 30 | 4.0 | 16.2 | 1,439 |
| 26† | 4.1 | 15.7 | 1,434 |
| 6 | 4.6 | 12.0 | 1,415 |
| 33 | 4.9 | 7.85 | 1,232 |
| 69 | 5.6 | 5.65 | 1,195 |
| 80 | 6.4 | 4.45 | 1,207 |
| 56 | 6.4 | 3.23 | 1,040 |
| 94 | 6.9 | 4.61 | 1,336 |
| 42† | 7.5 | 3.35 | 1,239 |
| 85 | 7.6 | 4.52 | 1,448 |
| 103 | 7.6 | 6.06 | 1,671 |
| 19† | 7.9 | 1.39 | 849 |
| 21† | 9.5 | 4.72 | 1,853 |
| 1 | 10.6 | 2.98 | 1,651 |
| 54† | 13.7 | 1.32 | 1,432 |
| Mean | 6.9 | 6.58 | 1,348 |
| Min | 3.8 | 1.32 | 849 |
| Max | 13.7 | 16.2 | 1,853 |
| SD | 2.7 | 4.73 | 252 |
Average viral load for a given patient was calculated as the arithmetic mean of all viral load measurements for that patient. This method presupposes nothing about the trend viral load follows over time, provided measurements are taken at roughly even time intervals, and is therefore compatible with both the set point hypothesis and observations of exponential increase over time. Note that the viral constant (ta·v̄0.49) is relatively unchanged from patient to patient around a mean of 1,348 ± 63 viral years (σ = 252). †, Patients who experienced a significant exponential increase in viral load.
Figure 1.

CD4+ cell counts and viral load measurements. CD4+ cell declines and viral load measurements for representative patients (a) 056 and (b) 042. For simplicity, the predicted survival time, tp, is taken as the time at which the CD4+ cell count falls to zero. Patient 042 (b) was one of six patients who experienced a statistically significant exponential increase in viral load; however, our method of calculating the average viral load, v̄, makes no assumptions about the trend viral load follows over time, provided viral load data points are taken at roughly equal intervals. Viral constants are calculated as in Table 1. ta, actual survival time.
As noted, several studies have found a general inverse relationship between viral load and survival time across groups of patients. We were interested in determining what form this inverse relationship takes on a patient-by-patient basis, using the average viral load, v̄, and the time from seroconversion until death, ta, to measure viral load and survival time (Table 1). Note that use of the average (arithmetic mean) of a given patient’s viral load measurements presupposes nothing about the trend viral load follows over time, provided that measurements are taken at roughly equal time intervals (see Discussion); hence this approach is compatible with both the set point hypothesis and observations of increase over time (10).
RESULTS
We plotted ta against v̄ on log-log axes and found a highly significant (P < 0.0001; geometric mean regression) negative correlation between viral load and survival time. The correlation was surprisingly tight considering the error inherent in seroconversion date estimation, explaining nearly 75% of the observed variation (r2 = 0.74). This observation suggests the relationship
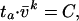 |
1 |
where k = 0.49 ± 0.07 and the constant C, the “viral constant,” equals 1,325 (Table 1) (16). This means, for example, that doubling a patient’s survival time requires a 4-fold reduction in average viral load.
We can interpret the relative uniformity (see Table 1) of the viral constant by saying that some function of the total amount of viremia experienced by a given patient from seroconversion until death is roughly constant. We coin the term “viral year” to describe this quantity; so, for example, a patient with an average viral load of 1 × 104 copies of HIV-1 RNA/ml who survived for 13 years would be said to have experienced 1,300 viral years.
It is clear from Fig. 2a that this relationship exhibits some variation. To better visualize it, we plotted a histogram of viral constants from these 16 patients (Fig. 3), which showed a distribution of values around a mean of 1,348 ± 63 viral years, with a SD of 252, consistent with the value of 1,325 for C given by the regression above. Interestingly, the only patient whose viral constant was below 1,000 (patient 19) was also the only patient whose viral load measurements were unevenly spaced. After a series of measurements approximately 8 months apart, viral load for this patient was not recorded for a period of 4 years (data not shown). Excluding this patient, the relationship in Fig. 2a now explained more than 82% of the observed variation without statistically changing k or C (data not shown). Hence even frequency of measurements may be important for accurate viral constant estimation.
Figure 2.

A simple relationship between viral load and survival time. Viral load and survival time can be described by a simple phenomenological relationship (a) that explains nearly 75% of the observed variation; most of the remaining variation can be explained by error associated with seroconversion date estimation (±1 year for this group). Underlining the importance of accurate seroconversion date estimation, relaxing selection criteria to include patients seropositive at the start of the study (b) resulted in a much looser fit; however, it changed neither C nor k and the relationship remained highly significant. This relationship may reflect the underlying dynamics of CD4+ cell decline, because using CD4+ cell decline to predict survival time (c) gives results similar to those seen in a and b. Better estimation of seroconversion dates is required to further evaluate this possibility.
Figure 3.

Viral constant is relatively constant from patient to patient. There is some variability around the value of the viral constant, most likely because of imprecise estimation of seroconversion dates. Interestingly, the only patient whose viral constant was below 1,000 (patient 19) was also the only patient whose viral load measurements were not roughly evenly spaced, making calculation of v̄ less accurate (see main text). Hence frequency of measurements is also important for accurate viral constant estimation.
It is likely that much of the variation in Fig. 2a may be the result of imprecision in estimating seroconversion dates, which could on average still account for a 13% error in survival time. To test this possibility, we relaxed our selection criteria to include patients who were seropositive at the start of the study (47 patients) and repeated the above analysis (Fig. 2b). The resulting plot still showed a highly significant correlation, with a slope in line with that of Fig. 2a, but one that now explained only 30% of the observed variation. Hence imprecision in estimating seroconversion dates indeed represents a significant source of error.
We next sought to link this relationship to the proximal cause of disease progression, CD4+ cell decline. Because we were interested in time course data, we first considered only patients with three or more CD4+ cell count measurements who exhibited a statistically significant CD4+ cell decline over time (40 patients). Following previous reports (17), we described this decline according to the equation
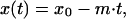 |
2 |
where x(t) is the CD4+ cell count at time t, xo is the baseline CD4+ cell count (taken at seroconversion), and m is the slope of CD4+ cell decline (Fig. 1).
Because progression to AIDS and death is associated with decreasing CD4+ cell counts, we made the approximation that a count of zero coincides with death. According to Eq. 2, this happens at time tp = xo/m, the predicted survival time (Fig. 1, arrows). We tested its predictive power by comparing actual (ta) and predicted (tp) survival times for the 25 of these 40 patients who progressed to AIDS and died during the study and found a significant correlation with regression slope of approximately 1 (significance of correlation P = 0.0001, t test for slope different from one P = 0.07, r2 = 0.70), supporting our use of tp.
A log-log plot of tp vs. average viral load v̄ again revealed a highly significant correlation (Fig. 2c), with a slope in close agreement with that of Fig. 2a. The relative looseness of the correlation is most likely caused by error in approximating tp and in estimation of seroconversion dates for those patients seropositive at the study’s inception. This finding suggests that the relationship summarized in Eq. 1 can be rewritten to include both baseline CD4+ cell count and rate of decline, although more precise data are required to further evaluate this possibility.
DISCUSSION
In this study we sought to quantitate the previously observed inverse relationship between viral load and survival time; we then asked whether this general observation might be a reflection of an underlying inverse relationship that holds for all patients, in the hope that this might help shed light on the mechanism of AIDS pathogenesis.
Symbolically, the view of HIV-1 disease progression that emerges from the present study is of the form
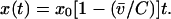 |
3 |
Note the inclusion of both rate of CD4+ cell decline, m, and CD4+ cell count at seroconversion, xo. This means that for a given baseline CD4+ cell count, higher viral load is associated with faster decline and shorter predicted survival time (Fig. 4a); but consistent with the view that viral load drives progression, it also means that for a given viral load, higher baseline CD4+ cell count does not change predicted survival time (Fig. 4b).
Figure 4.

Viral load, CD4+ cell count, and survival time. For a given seroconversion CD4+ cell count, higher viral load is associated with faster decline and shorter predicted survival time (a); but counterintuitively, for a given viral load, higher seroconversion CD4+ cell count does not change predicted survival time (b). The dependence on seroconversion CD4+ cell count can be overlooked if large groups of patients are studied instead of individuals.
This finding confirms the observation that CD4+ cell count is by itself a “grossly inaccurate” indicator of viral load (3). However, it also qualifies the previous report of an inverse relationship between viral load and rate of CD4+ cell decline (5), because our findings suggest such a relationship should hold only if baseline CD4+ cell counts are roughly equal. The probable explanation is that in the cited work, as in other previous studies (11), patients were sorted into large risk groups by level of viral load. Because each such group contained more than 100 patients, average baseline CD4+ cell count was roughly constant from group to group, which could have obscured any dependence on baseline CD4+ cell count.
From a clinical perspective, our results suggest that survival time is determined not only by a patient’s present level of viremia but also by a patient’s history with the disease. This finding is consistent with the idea that HIV-1 infection is progressively lymphodegenerative, and hence results in impaired CD4+ cell replenishment, as seen in patients undergoing effective antiviral therapy (18). Because a change in viral load late in the disease will come after the patient already has faced many viral years of HIV-1, our results imply that treatment begun in late-stage disease will be less beneficial than treatment begun early, in agreement with the conventional view of “hit early, hit hard” (19–21), although for different reasons (Fig. 5a). However, the decision to begin therapy depends on a variety of practical clinical considerations.
Figure 5.

Quantitating the effect of therapy. Treatment begun early is more beneficial than treatment begun late, consistent with “hit early, hit hard” dogma (a); also, even moderate (5- to 10-fold) long-term reduction of viral load can be more beneficial than more potent (100- to 10,000-fold) but transient treatment (b). Figure is for a patient with pretreatment v̄ = 20,000 copies HIV-1 RNA/ml; fold reduction is calculated as v̄/v̄T (see main text). Curves are obtained from the exact expression t* = 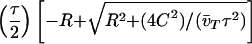 , where R = (v̄/v̄T) − 1, of which Eq. 4 is a series approximation.
, where R = (v̄/v̄T) − 1, of which Eq. 4 is a series approximation.
The quantitation summarized in Eq. 1 suggests that a given patient’s survival time is determined by the average viral load experienced by that patient over the course of infection. That our relationship explains nearly three-quarters of the observed variation in the data despite the significant error associated with seroconversion date estimation supports the conclusion that it holds across patients, and hence that it reflects the underlying mechanism of disease progression.
Our results are consistent with the view that progression is at some level quite simple. Upon seroconversion, a given patient can withstand only ≈1,300 viral years of HIV-1. Although there will likely be exceptions to this rule (which will fall in the tails of the distribution in Fig. 3), precision of the value for the viral constant will improve as more patients with more viral load measurements are analyzed.
The simplest interpretation is that viral load is a correlate of lymphocyte destruction. The more virus, the more rapidly lymphocytes are destroyed, the sooner the development of immunodeficiency, and consequently the faster the progression to AIDS and death. Assuming that productive CD4+ cell infection is proportional to the amount of stimulation by antigen-presenting cells, the value of k in Eq. 1 could be a consequence of the mechanics of antigen presentation. Four times as much virus leads to approximately twice as much CD4+ cell activation. This conclusion is consistent with a recent cross-sectional study that compared the activation state of CD4+ cells with viral load in a cohort of 30 HIV-1-infected patients (22).
That the viral constant is roughly unchanged from patient to patient then may reflect the fact that immune systems do not vastly differ from one person to another with respect to their CD4+ cell homeostatic mechanisms. Normal CD4+ homeostasis is maintained by a delicate balance between cell replenishment and cell death, which HIV-1 infection destroys (19, 23–30). Given evidence that the body responds to CD4+ cell depletion by maximizing replenishment (31), that cell count should decline at all implies that the rate of HIV-1-induced cell death is greater than the maximum rate of replenishment. Furthermore, because CD4+ cell decline is roughly linear, HIV-1-induced cell death must outstrip production by roughly the same amount at all times after seroconversion. Consideration of this point may help inform conjecture as to the mechanism of HIV-1 progression.
How does this finding relate to conventional wisdom regarding the viral set point (32)? If the set point for a given patient is a good approximation of the average viral load, predictions made from the two will be identical. However, this may not be the case in many patients, and the existence of a stable set point remains controversial (e.g., Fig. 1 a and b, Lower) (10, 33). Regardless, we note that our finding is compatible with the set point hypothesis, observations of increase in viral load (10), and observations that viral load fluctuates significantly over time (33). This compatibility is because the average will smooth out fluctuations over time, provided measurements are roughly evenly spaced.
Our quantitation suggests a new way to gauge the effects of a given regime of therapy. Consider a patient with average viral load v̄. According to Eq. 1, this patient’s predicted survival time will be t = C/v̄k. Assume treatment, initiated at time τ after seroconversion, reduces viral load to v̄T. The new predicted survival time, t*, then is approximated by the equation
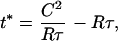 |
4 |
where R = (v̄/v̄T) − 1. In general, this means that even moderate (5- to 10-fold) long-term suppression of viral load is at least as beneficial as stronger (100- to 1,000-fold), but transient, treatment, as previously suggested (Fig. 5b) (34).
In sum, in the present work we have quantitated the previously observed relationship between viral load and survival time. The result provides evidence that disease progression in patients from across the clinical spectrum can be well described by a single simple phenomenological relationship. This, in turn, gives rise to a quantitative approach for assessing the benefits of antiviral therapy that may prove useful in evaluating mechanistic and mathematical models of AIDS pathogenesis (35, 36).
Acknowledgments
We thank R. May, V. Jansen, R. Ribeiro, and D. Callaway for helpful discussions and comments on the manuscript. We gratefully acknowledge the support of the Marshall Aid Commemoration Commission, United Kingdom (R.A.A.) and the Medical Research Council, United Kingdom (A.L.L.).
References
- 1.Haynes B F, Pantaleo G, Fauci A S. Science. 1996;271:324–328. doi: 10.1126/science.271.5247.324. [DOI] [PubMed] [Google Scholar]
- 2.Pantaleo G, Fauci A S. Annu Rev Microbiol. 1996;50:825–854. doi: 10.1146/annurev.micro.50.1.825. [DOI] [PubMed] [Google Scholar]
- 3.Mellors J W, Rinaldo C R, Jr, Gupta P, White R M, Todd J A, Kingsley L A. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien T R, Blattner W A, Waters D, Eyster E, Hilgartner M W, Cohen A R, Luban N, Hatzakis A, Aledort L M, Rosenberg P S, et al. J Am Med Assoc. 1996;276:105–110. [PubMed] [Google Scholar]
- 5.Mellors J W, Munoz A, Giorgi J V, Margolick J B, Tassoni C J, Gupta P, Kingsley L A, Todd J A, Saah A J, Detels R, et al. Ann Intern Med. 1997;126:946–954. doi: 10.7326/0003-4819-126-12-199706150-00003. [DOI] [PubMed] [Google Scholar]
- 6.Stein D S, Lyles R H, Graham N M, Tassoni C J, Margolick J B, Phair J P, Rinaldo C, Detels R, Saah A, Bilello J. J Infect Dis. 1997;176:1161–1167. doi: 10.1086/514108. [DOI] [PubMed] [Google Scholar]
- 7.de Wolf F, Spijkerman I, Schellekens P T, Langendam M, Kuiken C, Bakker M, Roos M, Coutinho R, Miedema F, Goudsmit J. AIDS. 1997;11:1799–1806. doi: 10.1097/00002030-199715000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Iuliano R, Forastieri G, Brizzi M, Mecocci L, Mazzotta F, Ceccherini-Nelli L. J Acquired Immune Defic Syndr Hum Retrovirol. 1997;14:408–414. doi: 10.1097/00042560-199704150-00003. [DOI] [PubMed] [Google Scholar]
- 9.Piatak M, Jr, Saag M S, Yang L C, Clark S J, Kappes J C, Luk K C, Hahn B H, Shaw G M, Lifson J D. Science. 1993;259:1749–1754. doi: 10.1126/science.8096089. [DOI] [PubMed] [Google Scholar]
- 10.O’Brien T R, Rosenberg P S, Yellin F, Goedert J J. J Acquired Immune Defic Syndr Hum Retrovirol. 1998;18:155–161. doi: 10.1097/00042560-199806010-00007. [DOI] [PubMed] [Google Scholar]
- 11.Touloumi G, Hatzakis A, Rosenberg P S, O’Brien T R, Goedert J J. AIDS. 1998;12:1691–1697. doi: 10.1097/00002030-199813000-00018. [DOI] [PubMed] [Google Scholar]
- 12.Sheppard H W, Lang W, Ascher M S, Vittinghoff E, Winkelstein W. AIDS. 1993;7:1159–1166. [PubMed] [Google Scholar]
- 13.Lang W, Perkins H, Anderson R E, Royce R, Jewell N, Winkelstein W., Jr J Acquired Immune Defic Syndr. 1989;2:63–69. [PubMed] [Google Scholar]
- 14.Henrard D R, Phillips J F, Muenz L R, Blattner W A, Wiesner D, Eyster M E, Goedert J J. J Am Med Assoc. 1995;274:554–558. [PubMed] [Google Scholar]
- 15.Goedert J J, Biggar R J, Melbye M, Mann D L, Wilson S, Gail M H, Grossman R J, DiGioia R A, Sanchez W C, Weiss S H, et al. J Am Med Assoc. 1987;257:331–334. [PubMed] [Google Scholar]
- 16.Sokal R R, Rohlf F J. Biometry: The Principles and Practice of Statistics in Biological Research. New York: Freeman; 1994. [Google Scholar]
- 17.Taylor J M G, Cumberland W G, Sy J P, Mitsuyasu R T. In: Models for Infectious Human Diseases: Their Structure and Relation to Data. Isham V, Medley G, editors. Cambridge: Cambridge Univ. Press; 1996. pp. 127–138. [Google Scholar]
- 18.Douek D C, McFarland R D, Keiser P H, Gage E A, Massey J M, Haynes B F, Polis M A, Haase A T, Feinberg M B, Sullivan J L, et al. Nature (London) 1998;396:690–695. doi: 10.1038/25374. [DOI] [PubMed] [Google Scholar]
- 19.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, et al. Nature (London) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 20.Bonhoeffer S, May R M, Shaw G M, Nowak M A. Proc Natl Acad Sci USA. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ribeiro R M, Bonhoeffer S, Nowak M A. AIDS. 1998;12:461–465. doi: 10.1097/00002030-199805000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Orendi J M, Bloem A C, Borleffs J C, Wijnholds F J, de Vos N M, Nottet H S, Visser M R, Snippe H, Verhoef J, Boucher C A. J Infect Dis. 1998;178:1279–1287. doi: 10.1086/314451. [DOI] [PubMed] [Google Scholar]
- 23.McMichael A. Cell. 1998;93:673–676. doi: 10.1016/s0092-8674(00)81428-2. [DOI] [PubMed] [Google Scholar]
- 24.Finzi D, Silliciano R F. Cell. 1998;93:665–671. doi: 10.1016/s0092-8674(00)81427-0. [DOI] [PubMed] [Google Scholar]
- 25.Mohri H, Bonhoeffer S, Monard S, Perelson A S, Ho D D. Science. 1998;279:1223–1227. doi: 10.1126/science.279.5354.1223. [DOI] [PubMed] [Google Scholar]
- 26.Rosenzweig M, DeMaria M A, Harper D M, Friedrich S, Jain R K, Johnson R P. Proc Natl Acad Sci USA. 1998;95:6388–6393. doi: 10.1073/pnas.95.11.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perelson A S, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho D D. Nature (London) 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 28.Chun T W, Carruth L, Finzi D, Shen X, DiGiuseppe J A, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn T C, et al. Nature (London) 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 29.Perelson A S, Neumann A U, Markowitz M, Leonard J M, Ho D D. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 30.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 31.McCune J M, Loftus R, Schmidt D K, Carroll P, Webster D, Swor-Yim L B, Francis I R, Gross B H, Grant R M. J Clin Invest. 1998;101:2301–2308. doi: 10.1172/JCI2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ho D D. Science. 1996;272:1124–1125. doi: 10.1126/science.272.5265.1124. [DOI] [PubMed] [Google Scholar]
- 33.Vidal C, Garcia F, Romeu J, Ruiz L, Miro J M, Cruceta A, Soriano A, Pumarola T, Clotet B, Gatell J M. AIDS. 1998;12:1285–1289. doi: 10.1097/00002030-199811000-00009. [DOI] [PubMed] [Google Scholar]
- 34.Coffin J M. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 35.Pilyugin S, Mittler J, Antia R. J Theor Biol. 1997;186:117–129. doi: 10.1006/jtbi.1996.0319. [DOI] [PubMed] [Google Scholar]
- 36.Nowak M A, Anderson R M, McLean A R, Wolfs T F, Goudsmit J, May R M. Science. 1991;254:963–969. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]