

Abstract
The human DINB1 gene shares a high degree of homology with the Escherichia coli dinB gene. Here, we purify the hDINB1-encoded protein and show that it is a DNA polymerase. Because hDinB1 is the eighth eukaryotic DNA polymerase to be described, we have named it DNA polymerase (Pol) θ. hPolθ is unable to bypass a cis-syn thymine–thymine dimer, nor does it bypass a (6–4) photoproduct or an abasic site. We also examine the fidelity of hPolθ on nondamaged DNA templates by steady-state kinetic analyses and find that hPolθ misincorporates deoxynucleotides with a frequency of about 10−3 to 10−4. We discuss the relationship between the fidelity of hPolθ and its inability to bypass DNA damage.
Unrepaired lesions in template DNA can block the replication machinery. Cells have evolved specialized polymerases to carry out translesion DNA synthesis in which nucleotides are incorporated opposite the lesion and subsequently extended from. Because many lesions are noncoding, incorrect nucleotides are often incorporated, which can lead to mutations. Recently, a new family of DNA polymerases has been identified; all of the members of this family contain five highly conserved motifs, I–V, and several of these polymerases participate in lesion bypass (1). In Escherichia coli, mutagenic translesion DNA synthesis is coordinated by the UmuD′2C complex (2, 3). UmuC, a member of this superfamily, has a weak DNA polymerase (Pol) activity (Pol V) that is greatly stimulated by UmuD′, RecA, and SSB (4, 5). The E. coli dinB gene, also a member of this superfamily, is required for untargeted mutagenesis in unirradiated λ phage (6), and it encodes Pol IV (7). As of yet, E. coli DinB protein has not been shown to participate in translesion DNA synthesis. Overexpression of DinB in E. coli results in an 1,000-fold increase of spontaneous −1 frameshifts (8), and purified Pol IV can efficiently extend from a misaligned primer-template (7).
In the yeast Saccharomyces cerevisiae, the REV1, REV3, and REV7 genes are required for DNA damage-induced mutagenesis. The Rev3 and Rev7 proteins form Pol ζ, which can weakly bypass a cis-syn thymine–thymine (T–T) dimer (9), and the Rev1 protein has a deoxycytidyltransferase activity (10). Although Rev1 contains all five of the conserved motifs (1), it lacks a bona fide polymerase activity and it preferentially inserts a dCMP residue across from an abasic site in DNA, which can then be extended by Pol ζ (10).
The S. cerevisiae RAD30 gene is involved in error-free bypass of UV-induced DNA damage, because mutations in it result in increased UV mutability (11, 12). The RAD30-encoded Pol η efficiently bypasses a cis-syn T–T dimer in template DNA, and it does so correctly by inserting two adenines across from the two thymines of the dimer (13). The human counterpart of yeast RAD30, hRAD30A, encodes the human Pol η, which can also bypass a cis-syn T–T dimer (14, 15). Patients with the variant form of xeroderma pigmentosum are defective in hPolη (14, 15), and as a consequence, they suffer from a high incidence of UV-induced skin cancers.
In the search for the human counterpart of yeast RAD30, in addition to the hRAD30A gene, we identified a second cDNA that encodes a Rad30-like protein. This gene is identical to the recently reported human DINB1 gene (16). Here, we purify the hDinB1 protein and show that it is a DNA polymerase. Because hDinB1 is the eighth eukaryotic DNA polymerase to be described, we have named it Polθ. We find that hPolθ does not bypass a cis-syn T–T dimer, a (6–4) photoproduct, or an abasic site. We determine the fidelity of hPolθ by measuring the steady-state kinetics of deoxynucleotide incorporation on undamaged DNA templates and show that it misincorporates deoxynucleotides with a frequency of about 10−3 to 10−4.
Materials and Methods
Cloning of hDinB1.
To identify the hDINB1 gene, we searched the database of expressed sequence tags (Dbest) for human cDNAs that share homology with the yeast RAD30 gene. One clone, accession no. AA576919 (536 bp), was identified; its encoded protein shared a high degree of homology with the E. coli DinB protein. The entire hDINB1 clone was subsequently derived from a high-throughput cDNA screening service (Genome Systems, St. Louis). A 291-bp PCR fragment which corresponds to nt +313 to +602 of the hDINB1 gene, was used to screen a human fetal brain cDNA library. One clone, 22054, was isolated and found to carry a 3.7-kb insert in plasmid pCDNA2.1 (Invitrogen).
DNA Sequencing.
The entire insert of clone 22054 was sequenced by using the Thermo Sequenase kit (Amersham Pharmacia), and confirmed from sequence analysis of cDNAs generated by reverse transcription–PCR from total HeLa RNA.
Generation of the D198E199 → N198Q199 Mutation.
The hDinB1 N198Q199 mutation was generated by PCR with the mutagenic oligonucleotide N6148 (5′-CAATTTTATGGCCATGAGTCTTAATCAAGCCTACTTGAAT-3′), and oligonucleotide LP78 (5′-GGTGACTCCATTTCCTTTCTGATCG-3′). The amplified 1.16-kb DNA fragment was digested with MscI and XbaI and used to replace the wild-type fragment in plasmid pBJ733 (see below). The resulting plasmid, pBJ752, was sequenced to confirm the presence of the mutations. No other mutations were found in the PCR fragment.
Expression of hDinB1 and Mutant Proteins.
To express the wild-type hDinB1 protein, the entire hDINB1 gene was amplified from clone 22054 by PCR by using the oligonucleotides: LP080 (5′-CCTGGGTACCGGATCCACATATGGATAGCACAAAGGAGAAGTGTG-3′) and LP079 (5′-CCGAGGTCGACGGATCCTGAGGAAGGATTATTGCACTTGCC-3′), which generate BamHI restriction sites 6 nt upstream and 128 nt downstream, respectively, of the hDINB1 ORF, and the resulting fragment was cloned into YIplac211. The SacI/SpeI DNA fragment encompassing nucleotides +157 to +2,468 in the 3′ nontranslated region was then replaced with wild-type DNA from clone 22054, generating plasmid pBJ733. All PCR-generated fragments were sequenced and found to contain no mutations. The 2.7-kb BamHI fragment containing the wild-type hDINB1 gene was then cloned in-frame with the glutathione S-transferase (GST) gene under the control of a galactose-inducible phosphoglycerate kinase promoter in the overexpression plasmid pHQ241, generating plasmid pBJ736. The hdinB1 N198Q199 mutant gene in pBJ752 was similarly cloned into pHQ241, generating plasmid pBJ754. Yeast strain BJ5464 harboring either pBJ736 or pBJ754 was grown in synthetic complete medium lacking Leu and containing 2% dextrose/3% glycerol/2.5% lactose overnight at 30°C. Cells were diluted 1:30 in synthetic complete medium lacking Leu as above but without dextrose and allowed to grow for 16 h before the addition of 2% galactose. Cells were grown for an additional 7 h before harvesting by centrifugation and then were stored at −70°C.
Purification of hPolθ.
To purify GST-hPolθ protein, yeast cells were resuspended in 2 ml of cell breakage buffer (50 mM Tris⋅HCl, pH 7.5/10% sucrose/300 mM NaCl/1 mM EDTA/0.5 mM benzamidine/0.5 mM PMSF/10 mM 2-mercaptoethanol/5 μg/ml aprotinin/5 μg/ml chymostatin/5 μg/ml pepstatin A/5 μg/ml leupeptin) per g of cells and lysed in a French press before centrifugation at 100,000 × g. The extract was then passed over a 100-μl glutathione Sepharose 4B column (Amersham Pharmacia) at 4°C, and the column was washed with 10 volumes of cell breakage buffer containing 1 M NaCl followed by equilibration in elution buffer lacking glutathione. GST-hPolθ was batch eluted with one column volume elution buffer (100 mM Tris⋅HCl, pH 8.0/100 mM NaCl/10% glycerol/0.01% Nonidet P-40/25 mM glutathione) three times at 4°C. Batches were pooled, dialyzed against 1,000 volumes KB buffer, and GST-hPolθ-containing fractions were aliquoted and frozen at −70°C.
DNA Polymerase Assays.
The standard polymerase reaction (10 μl) contained 25 mM KPO4 (pH 7.0)/5 mM MgCl2/5 mM dithiolthreitol/100 μg/ml BSA/10% glycerol/100 μM of each deoxynucleotide (dGTP, dATP, dTTP, and dCTP) except where noted/10 nM of 5′-32P-labeled oligonucleotide primer annealed to an oligonucleotide DNA template/0.5 nM GST-hPolθ. Reactions were terminated by the addition of 50 mM EDTA and DNA products were precipitated with six volumes of ice-cold acetone. Samples were dried under vacuum, resuspended in loading buffer (95% formamide/0.3% cyanol blue/0.3% bromophenol blue) and heat denatured at 95°C before resolving products on 10% polyacrylamide gels containing 8 M urea. Gels were dried before autoradiography at −70°C.
DNA Substrates.
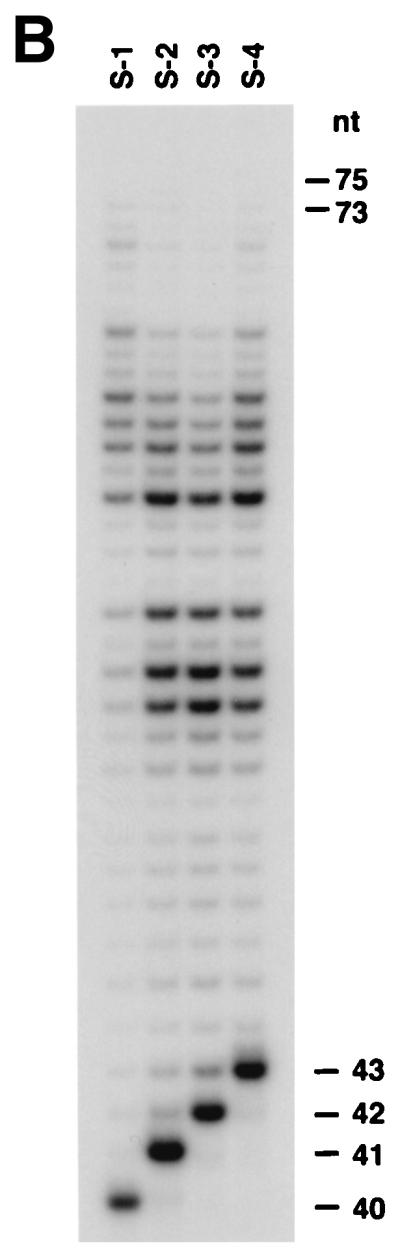
Substrates S-1 (template C), S-2 (template G), S-3 (template T), and S-4 (template A) were generated by annealing the 75 mer template oligonucleotide (5′-AGCTACCATGCCTGCCTCAAGAGTTCGTAACATGCCTACACTGGAGTACCGGAGCATCGTCGTGACTGGGAAAAC-3′) to 5′-32P-radiolabeled primer oligonucleotides: N4264, 5′-GTTTTCCCAGTCACGACGATGCTCCGGTACTCCAGTGTAG-3′; N4265, 5′-GTTTTCCCAGTCACGACGATGCTCCGGTACTCCAGTGTAGG-3′; N4266, 5′-GTTTTCCCAGTCACGACGATGCTCCGGTACTCCAGTGTAGGC-3′; and N4267, 5′-GTTTTCCCAGTCACGACGATGCTCCGGTACTCCAGTGTAGGCA-3′, respectively. For damage bypass assays, the “running start” oligonucleotide N4264 was annealed to the 75 mer template (5′-AGCTACCATGCCTGCACGAAGAGTTCGTATTATGCCTACACT- GGAGTACCGGAGCATCGTCGTGACTGGGAAAAC-3′) containing either a cis-syn T–T dimer, a (6–4) T–T photoproduct, or no damage at the underlined position, which were derived from the 10mer oligonucleotide 5′-CGTATTATGC-3′ by ligation to flanking 25 mer and 40 mer oligonucleotides. The modified oligonucleotide 10 mer containing the site-specific T–T dimer or a (6–4) T–T photoproduct were generated and purified as described (17, 18). The position of the abasic site corresponds to the 3′ thymine in the nondamaged 75 mer template. To assay for frameshifting, the oligonucleotides LP108 (5′-GTTTTCCCAGTCACGACGGG-3′), LP109 (5′-GTTTTCCCAGTCACGACGGC-3′), or LP107 (5′-GTTTTCCCAGTCACGACGGT-3′) were annealed to the templates N6146 (5′-AGCTACCATGCCTGCCGGGGCCCGTCATGACTGGGAAAAC-3′) or N6147 (5′-AGCTACCATGCCTGCGGGGCCCCGTCATGACTGGGAAAAC-3′).
DNA Polymerase Fidelity Measurements.
DNA polymerase fidelity was calculated from deoxynucleotide incorporation kinetics as described (19, 20). The standard DNA polymerase assay was used except that the reaction volume was reduced to 5 μl and only a single deoxynucleotide was included. The concentrations of deoxynucleotides were varied from 0 to 25 μM for the correct dNTP and from 0 to 2,000 μM for the incorrect dNTPs. Substrates S-1, S-2, S-3, and S-4 containing the template C, G, T, or A residues, respectively, are described above. Gel band intensities of the substrate and products of the deoxynucleotide incorporation reactions were quantitated by using a PhosphorImager and the imagequant software (Molecular Dynamics). For each concentration of dNTP, the observed rate of deoxynucleotide incorporation, vobs, was determined by dividing the amount of product formed by the reaction time. The vobs was then plotted as a function of the dNTP concentration and the data were fit to the Michaelis–Menten equation describing a hyperbola (Eq. 1): vobs = (Vmax × [dNTP])/(Km + [dNTP]). From the best fit curve, the apparent Km and Vmax steady-state kinetic parameters for the incorporation of both correct and incorrect deoxynucleotides were obtained and these parameters were used to calculate the frequency of deoxynucleotide misincorporation, finc, by using the following equation (Eq. 2): finc = (Vmax/Km)incorrect/(Vmax/Km)correct.
Results
To identify the human genes related to yeast RAD30, we searched the database of human expressed sequence tagged cDNAs and identified a clone, AA576919, that bears significant homology to yeast RAD30. This cDNA sequence was used to screen a human fetal brain cDNA library, and a single clone, 22054, containing a 3.7-kb insert was isolated. Sequence analysis revealed a single ORF that encodes a protein highly homologous to the E. coli DinB protein. While this work was in progress, the sequences of the human and mouse DINB1 genes (accession nos. AF163570 and AF163571, respectively) were reported (16); our hDINB1 sequence is identical to the reported sequence.
To determine whether hDINB1 encodes a DNA polymerase, we purified the encoded protein. To accomplish this, we fused the hDINB1 gene in-frame with the GST gene that is under the control of a galactose-inducible phosphoglycerate kinase promoter. The resulting fusion protein was purified from a protease-deficient strain of yeast (Fig. 1). The hDinB1 protein was assayed for the incorporation of each deoxynucleotide opposite its complement in template DNA. hDinB1 is able to incorporate all four nucleotides in a template-dependent fashion (Fig. 2A), and in the presence of all four deoxynucleotides, hDinB1 synthesizes DNA nearly to the end of the template (Fig. 2B). Interestingly, whereas most DNA polymerases synthesize to the end of template DNA, or even add an additional residue, hDinB1 protein stops synthesis one to two nucleotides before the end of the template.
Figure 1.
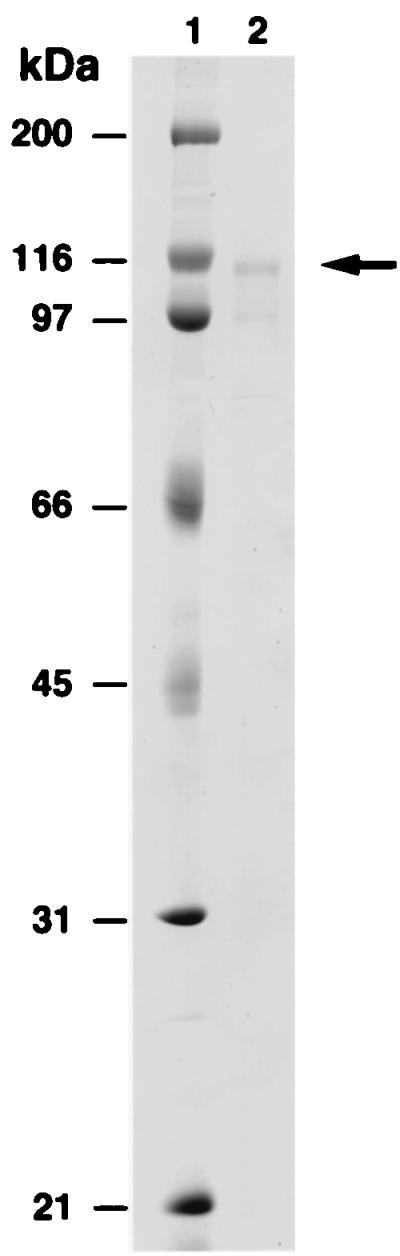
Purified GST-hDinB1 protein. Lane 1, molecular weight markers and lane 2, 100 ng hDinB1 protein. The arrow on the right indicates the position of the full-length GST-hDinB1 protein. The lower band is a degradation product of this protein.
Figure 2.

Deoxynucleotide incorporation and DNA polymerase activity of hDinB1. (A) A 0.5 nM concentration of hDinB1 protein was incubated with 10 nM DNA substrate and with a 10 μM concentration of a single deoxynucleotide as indicated. A portion of each substrate is shown on top. (B) A 0.5 nM concentration of hDinB1 protein was incubated with 10 nM DNA substrate S-1, S-2, S-3, or S-4 in the presence of a 100 μM concentration of each of the four dNTPs. Sizes of primers and products are shown on the right.
To ensure that the observed DNA polymerase activity was intrinsic to the hDinB1 protein, we generated a mutant hDinB1 protein and showed that it lacked the DNA polymerase activity. The highly conserved Asp and Glu residues in motif III of Rad30-related proteins are present at positions 198 and 199 in the hDinB1 protein, respectively, and these were changed to Asn and Gln, respectively. The mutant protein was purified from yeast in the same manner as the wild-type hDinB1 protein. This mutant protein displayed the same chromatographic properties as the wild-type protein, but it lacked any DNA polymerase activity (data not shown). Thus, the observed DNA polymerase activity is intrinsic to hDinB1 protein. Because hDINB1 encodes the eighth eukaryotic DNA polymerase, we have named it polymerase θ (Polθ).
To determine whether hPolθ can bypass DNA lesions, we examined the ability of this protein to replicate from templates containing a cis-syn T–T dimer, a (6–4) T–T photoproduct, or an abasic site. hPolθ is unable to bypass a cis-syn T–T dimer, and moreover, synthesis by hPolθ was strongly blocked two nucleotides before the dimer (Fig. 3). hPolθ also does not bypass a (6–4) photoproduct or an abasic site (Fig. 3).
Figure 3.

Lack of damage bypass by hPolθ; 0.5 nM hPolθ was incubated with 10 nM DNA substrate in the presence of a 100 μM concentration of each of the four dNTPs. Lane 1, undamaged DNA; lane 2, a cis-syn T–T dimer containing template; lane 3, a (6–4) T–T photoproduct containing template; and lane 4, an abasic site containing template. The arrow on the right indicates the position of the first T of the dimer, the first T of the (6–4) photoproduct, or the position of the abasic site in the template.
Whereas the E. coli DinB protein does not readily extend a mispaired 3′ terminus, it can extend from a misaligned primer–template (7). To determine whether hDinB1 has a similar property, we examined DNA synthesis from DNA substrates that can misalign, and the various DNA substrates used are shown in Fig. 4A. The G:C DNA substrate has a correct G:C bp at the 3′ terminus of the primer. The C:C (−1) substrate has a C:C mispair at the primer terminus; however, this C in the primer can misalign with the nucleotide G at position 21 in the template. Extension from the misaligned primer would result in a product one nucleotide shorter than full length. The C:C (−2) substrate also has a C:C mispair at the primer–terminus, and in this case, this C in the primer can misalign with a G residue at position 22 in the template, extension from which would result in a product 2 nt shorter than full length. Finally, the substrate with a T:C mispair at the primer terminus cannot misalign on the template. Because of the presence of the proofreading 3′ to 5′ exonuclease activity, the Klenow fragment removes the mispair and synthesizes full-length DNA from the mispaired substrates (Fig. 4B, lanes 2–4). However, even though hPolθ has no proofreading 3′ to 5′ exonuclease activity, it can synthesize DNA from the C:C (−1) substrate by misaligning (Fig. 4B, lane 6), but it is very inefficient at extending from the C:C (−2) substrate (Fig. 4B, lane 7), and it extends weakly from the T:C substrate (Fig. 4B, lane 8). To examine whether the ability to extend from the misaligned or mispaired substrates was unique to hPolθ, we examined the ability of hPolη to extend from these different primer termini. hPolη, which also lacks a proofreading 3′ to 5′ exonuclease activity, is able to synthesize from the C:C (−1) misaligned substrate (Fig. 4B, lane 10); and moreover, it is more efficient than hPolθ in synthesizing from the C:C (−2) misaligned substrate (Fig. 4B, lane 11). hPolη is also able to synthesize from the mispaired T:C substrate (Fig. 4B, lane 12).
Figure 4.

Synthesis from misaligned or mispaired primer–template substrates by hPolθ. (A) A portion of each primer–template substrate is shown. In each case, the primer is 20 nt long and the template is 40 nt long. The G:C substrate contains the correct bp at the primer–template junction, and extension would result in full-length product. In the C:C (−1) substrate, a C:C mispair is at the primer–template junction. The C in the primer, however, can base pair with the underlined G residue at position 21 in the template, extension from which would result in a −1 frameshift. The C:C (−2) substrate also has a C:C mispair at the primer terminus, and in this case, the C in the primer can base pair with the underlined G residue at position 22 in the template. Synthesis from this misaligned template would result in a −2 frameshift. The T:C substrate is unable to misalign; therefore, synthesis from this primer would occur by extension from the mispair and result in full-length product. (B) Extension of G:C, C:C (−1), C:C (−2), and T:C DNA substrates by E. coli Klenow fragment, hPolθ, or hPolη. Lanes 1–4, extension of each substrate by Klenow fragment; lanes 5–8, extension of each substrate by hPolθ; and lanes 9–12, extension of each substrate by hPolη. Lengths of primer extension products are indicated on the left.
To determine the fidelity of hPolθ, we used a steady-state kinetics assay to measure the incorporation of correct and incorrect deoxynucleotides opposite each of the four template nucleotides. hPolθ was incubated with primer–template DNA substrates and increasing concentrations of correct or incorrect deoxynucleotides. DNA products were separated by gel electrophoresis. The pattern of incorporation of G, A, T, and C opposite the template C residue is shown in Fig. 5A, whereas the incorporation of G, A, T, and C opposite the template G is shown in Fig. 5B. The amount of deoxynucleotide incorporation was quantified by phosphorimaging, and from the kinetics of deoxynucleotide incorporation, the steady-state Vmax and apparent Km parameters were obtained from the curve fit to Eq. 1 (see Materials and Methods). The frequency of misincorporation, finc, for each incorrect nucleotide across from the respective template nucleotide was then obtained from Eq. 2 (see Materials and Methods). For instance, for the incorporation of G opposite the template C, the Vmax and apparent Km values are 1.9 nM/min and 0.6 μM, respectively, and for the incorporation of C opposite the template C, the Vmax and apparent Km values are 1.2 nM/min and 346 μM, respectively; thus, from Eq. 2, the finc for misincorporating C opposite C is 9.4 × 10−4 (Table 1). The Vmax, Km, and finc values for the incorporation of all four deoxynucleotides opposite each template nucleotide are shown in Table 1. Overall, the fidelity of hPolθ ranged from 1.5 × 10−4 for the misincorporation of T opposite template T to 3.0 × 10−3 for the misincorporation of G opposite template A.
Figure 5.

Steady-state deoxynucleotide incorporation by hPolθ. (A) Incorporation of the correct and the incorrect deoxynucleotides across from a template C residue in substrate S-1. A portion of the substrate is shown on top. (B) Incorporation of the correct and the incorrect deoxynucleotides across from a template G residue in substrate S-2. Deoxynucleotide concentrations (μM) are indicated.
Table 1.
Steady-state kinetic values and misincorporation frequencies for hPolθ
| Template residue | Incoming dNTP | Vmax, nM/min | Km, μM | Vmax/Km | finc |
|---|---|---|---|---|---|
| G | G | 0.3 ± 0.02 | 284 ± 47 | 0.001 | 2.2 × 10−4 |
| A | 0.7 ± 0.04 | 178 ± 37 | 0.004 | 8.9 × 10−4 | |
| T | 0.8 ± 0.04 | 638 ± 88 | 0.001 | 2.2 × 10−4 | |
| C | 1.8 ± 0.06 | 0.4 ± 0.05 | 4.5 | ||
| A | G | 0.3 ± 0.02 | 118 ± 41 | 0.003 | 3.0 × 10−3 |
| A | 0.2 ± 0.01 | 350 ± 56 | 6 × 10−4 | 6.0 × 10−4 | |
| T | 2.2 ± 0.3 | 2.2 ± 0.8 | 1.0 | ||
| C | 1.2 ± 0.1 | 1,420 ± 305 | 8 × 10−4 | 8.0 × 10−4 | |
| T | G | 0.38 ± 0.05 | 117 ± 33 | 0.003 | 4.4 × 10−4 |
| A | 1.9 ± 0.05 | 0.28 ± 0.09 | 6.8 | ||
| T | 0.3 ± 0.02 | 213 ± 55 | 0.001 | 1.5 × 10−4 | |
| C | 1.0 ± 0.02 | 551 ± 33 | 0.002 | 2.9 × 10−4 | |
| C | G | 1.9 ± 0.08 | 0.6 ± 0.1 | 3.2 | |
| A | 0.22 ± 0.01 | 251 ± 31 | 9 × 10−4 | 2.8 × 10−4 | |
| T | 0.07 ± 0.01 | 121 ± 41 | 6 × 10−4 | 1.9 × 10−4 | |
| C | 1.2 ± 0.4 | 346 ± 37 | 0.003 | 9.4 × 10−4 |
Discussion
Whereas UmuC-like proteins have been identified in bacterial species, Rad30 and Rev1 types of proteins exist only in eukaryotes, and DinB-like proteins have been identified in both prokaryotes and eukaryotes (1). A greater diversity of these proteins seems to be present in higher eukaryotes, because humans contain two Rad30-like proteins, a DinB-like protein, and a Rev1 cognate, whereas S. cerevisiae has only the Rad30 and Rev1 proteins (1). The S. cerevisiae RAD30 and the human hRAD30A genes encode Polη which bypasses a cis-syn T–T dimer by inserting two adenines across from the dimer, and consistent with this biochemical observation, mutations in this gene in both yeast and humans cause UV hypermutability, and defects in hRAD30A in humans result in the cancer-prone variant form of xeroderma pigmentosum (14, 15). The protein encoded by hRAD30B more closely resembles hRad30A than it does hDinB1 (16, 21).
Here, we purify the human DinB1 protein and show that it is a DNA polymerase, which we have named Polθ. The sequence of the protein encoded by the Drosophila mus308 gene shares similarity with the polymerase domain of E. coli DNA polymerase I (22), and a sequence related to mus308 has also been identified in humans. Based on this sequence similarity, the encoded human protein was named Polη by one group (23) and Polθ by another (24). However, because no DNA polymerase activity has been identified for either of these Drosophila or human proteins, we consider this nomenclature premature.
In E. coli, dinB affects untargeted mutagenesis in phage λ, which is observed when undamaged λ DNA infects UV-irradiated E. coli cells. Also, overexpression of dinB in the absence of DNA-damaging agents enhances the mutagenesis of F′ lac plasmids, and results in a predominant increase in the frequency of −1 frameshifts in homopolymeric runs. Purified E. coli DinB protein can synthesize DNA from a G-G mismatch placed within a short repetitive sequence, but it does so by misaligning the primer and generating −1 frameshifts (7). A role for the E. coli dinB gene in the replication of repetitive DNA sequences via template misalignment has been suggested (7). However, we find that even though hPolθ can elongate from a C:C (−1) misaligned primer, it is no more efficient at this than is hPolη. Moreover, hPolθ is quite inefficient at elongating from a template–primer where replication would occur by misalignment that would generate a deletion of two nucleotides, whereas hPolη can extend from such a substrate. These results suggest that hPolθ may not be specifically involved in the replication of repetitive sequences via template misalignment, as has been proposed for the E. coli DinB protein.
Our results indicate that hPolθ is unable to bypass a cis-syn T–T dimer, a (6–4) T–T photoproduct, or an abasic site. The inability of hPolθ to bypass a T–T dimer is in contrast to the efficient bypass of this lesion by yeast and human Polη. To understand the probable basis for this difference in the ability of Polη vs. Polθ to bypass DNA lesions, we determined the fidelity of hPolθ by measuring the steady-state kinetics of correct and incorrect deoxynucleotide incorporation across from the nondamaged DNA templates. Our studies indicate that hPolθ misincorporates deoxynucleotides with a frequency of 10−3 to 10−4. In contrast, the fidelity of yeast and human Polη is 10−2 to 10−3 (25, 26). As discussed below, fidelity may be a key factor in determining whether or not a DNA polymerase will bypass a DNA lesion.
Most DNA polymerases, but especially those involved in replication, misincorporate deoxynucleotides very infrequently (19, 20, 27, 28). This high fidelity derives, in part, from the intolerance of the active site of these enzymes for geometric distortions in DNA (27), and unless the geometry is correct, they do not form a phosphodiester bond between the primer and the incoming deoxynucleotide (29). Thus, even though the thymines of a T–T dimer can base pair with adenines (30, 31), a dimer is a block to most DNA polymerases, presumably because the active site of these enzymes is unable to deal with the DNA distortion caused by the dimer (32, 33). The ability of Polη to bypass a T–T dimer would suggest that relative to other DNA polymerases, the active site of Polη is more flexible, and that renders the enzyme more tolerant of DNA distortions. That, in turn, would be expected to lower the fidelity, and consistent with this, Polη is a low-fidelity enzyme (25, 26). In contrast, the eukaryotic replicative Polδ has an error rate of ≈10−5, and Polα, required for lagging strand DNA synthesis, has an error rate of 10−3 to 10−4 (34). Thus, the error rate of hPolθ is about the same as that for Polα. We note that none of these DNA polymerases, Polδ, Polα, or hPolθ, which have error rates of 10−3 to 10−5, bypass a T–T dimer (refs. 9 and 13, and this study), whereas yeast and hPolη, which have error rates of 10−2 to 10−3, are able to do so. Thus, a low-fidelity enzyme may be better able to bypass DNA lesions than an enzyme with a high fidelity.
Acknowledgments
We thank Christine Kondratick for help with cloning of the hDINB1 gene, and Richard Hodge for providing the thymine dimer and (6–4) photoproduct-containing DNA, the construction of which was supported by National Institute on Environmental Health Sciences Center Grant P30-ESO6676. This work was supported by National Institutes of Health Grant GM19261.
Abbreviations
- Pol
DNA polymerase
- T–T dimer
thymine–thymine dimer
- GST
glutathione S-transferase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Johnson R E, Washington M T, Prakash S, Prakash L. Proc Natl Acad Sci USA. 1999;96:12224–12226. doi: 10.1073/pnas.96.22.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang M, Bruck I, Eritja R, Turner J, Frank E G, Woodgate R, O'Donnell M, Goodman M F. Proc Natl Acad Sci USA. 1998;95:9755–9760. doi: 10.1073/pnas.95.17.9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reuven N B, Tomer G, Livneh Z. Mol Cell. 1998;2:191–199. doi: 10.1016/s1097-2765(00)80129-x. [DOI] [PubMed] [Google Scholar]
- 4.Tang M, Shen X, Frank E G, O'Donnell M, Woodgate R, Goodman M F. Proc Natl Acad Sci USA. 1999;96:8919–8924. doi: 10.1073/pnas.96.16.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reuven N B, Arad G, Maor-Shoshani A, Livneh Z. J Biol Chem. 1999;274:31763–31766. doi: 10.1074/jbc.274.45.31763. [DOI] [PubMed] [Google Scholar]
- 6.Brotcorne-Lannoye A, Maenhaut-Michel G. Proc Natl Acad Sci USA. 1986;83:3904–3908. doi: 10.1073/pnas.83.11.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner J, Gruz P, Kim S-R, Yamada M, Matsui K, Fuchs R P P, Nohmi T. Mol Cell. 1999;4:281–286. doi: 10.1016/s1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- 8.Kim S-R, Maenhaut-Michel G, Yamada M, Yamamoto Y, Matsui K, Sofuni T, Nohmi T, Ohmori H. Proc Natl Acad Sci USA. 1997;94:13792–13797. doi: 10.1073/pnas.94.25.13792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson J R, Lawrence C W, Hinkle D C. Science. 1996;272:1646–1649. doi: 10.1126/science.272.5268.1646. [DOI] [PubMed] [Google Scholar]
- 10.Nelson J R, Lawrence C W, Hinkle D C. Nature (London) 1996;382:729–731. doi: 10.1038/382729a0. [DOI] [PubMed] [Google Scholar]
- 11.McDonald J P, Levine A S, Woodgate R. Genetics. 1997;147:1557–1568. doi: 10.1093/genetics/147.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson R E, Prakash S, Prakash L. J Biol Chem. 1999;274:15975–15977. doi: 10.1074/jbc.274.23.15975. [DOI] [PubMed] [Google Scholar]
- 13.Johnson R E, Prakash S, Prakash L. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- 14.Johnson R E, Kondratick C M, Prakash S, Prakash L. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 15.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. Nature (London) 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 16.Gerlach V L, Aravind L, Gotway G, Schultz R A, Koonin E V, Friedberg E C. Proc Natl Acad Sci USA. 1999;96:11922–11927. doi: 10.1073/pnas.96.21.11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banerjee S K, Christensen R B, Lawrence C W, LeClerc J E. Proc Natl Acad Sci USA. 1988;85:8141–8145. doi: 10.1073/pnas.85.21.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LeClerc J E, Borden A, Lawrence C W. Proc Natl Acad Sci USA. 1991;88:9685–9689. doi: 10.1073/pnas.88.21.9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodman M F, Creighton S, Bloom L B, Petruska J. Crit Rev Biochem Mol Biol. 1993;28:83–126. doi: 10.3109/10409239309086792. [DOI] [PubMed] [Google Scholar]
- 20.Creighton S, Bloom L B, Goodman M F. Methods Enzymol. 1995;262:232–256. doi: 10.1016/0076-6879(95)62021-4. [DOI] [PubMed] [Google Scholar]
- 21.McDonald J P, Rapic-Otrin V, Epstein J A, Broughton B C, Wang X, Lehmann A R, Wolgemuth D J, Woodgate R. Genomics. 1999;60:20–30. doi: 10.1006/geno.1999.5906. [DOI] [PubMed] [Google Scholar]
- 22.Harris P V, Mazina O M, Leonhardt E A, Case R B, Boyd J B, Burtis K C. Mol Cell Biol. 1996;16:5764–5771. doi: 10.1128/mcb.16.10.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burtis K C, Harris P V. Curr Biol. 1997;7:R743–R744. doi: 10.1016/s0960-9822(06)00391-5. [DOI] [PubMed] [Google Scholar]
- 24.Sharief F S, Vojta P J, Ropp P A, Copeland W C. Genomics. 1999;59:90–96. doi: 10.1006/geno.1999.5843. [DOI] [PubMed] [Google Scholar]
- 25.Washington M T, Johnson R E, Prakash S, Prakash L. J Biol Chem. 1999;274:36835–36838. doi: 10.1074/jbc.274.52.36835. [DOI] [PubMed] [Google Scholar]
- 26.Johnson R E, Washington M T, Prakash S, Prakash L. J Biol Chem. 2000;275:7447–7450. doi: 10.1074/jbc.275.11.7447. [DOI] [PubMed] [Google Scholar]
- 27.Echols H, Goodman M F. Annu Rev Biochem. 1991;60:477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 28.Goodman M F, Fygenson D K. Genetics. 1998;148:1475–1482. doi: 10.1093/genetics/148.4.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steitz T A. J Biol Chem. 1999;274:17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 30.Kemmink J, Boelens R, Koning T, van der Marel G A, van Boom J H, Kaptein R. Nucleic Acids Res. 1987;15:4645–4653. doi: 10.1093/nar/15.11.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J-K, Patel D, Choi B-S. Photochem Photobiol. 1995;62:44–50. doi: 10.1111/j.1751-1097.1995.tb05236.x. [DOI] [PubMed] [Google Scholar]
- 32.Ciarrocchi G, Pedrini A M. J Mol Biol. 1982;155:177–183. doi: 10.1016/0022-2836(82)90445-4. [DOI] [PubMed] [Google Scholar]
- 33.Wang C-I, Taylor J-S. Proc Natl Acad Sci USA. 1991;88:9072–9076. doi: 10.1073/pnas.88.20.9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas D C, Roberts J D, Sabatino R D, Myers T W, Tan C-K, Downey K M, So A G, Bambara R A, Kunkel T A. Biochemistry. 1991;30:11751–11759. doi: 10.1021/bi00115a003. [DOI] [PubMed] [Google Scholar]