

Abstract
We have noted consistent structural similarities among unrelated proteases. In comparison with other proteins of similar size, proteases have smaller than average surface areas, smaller radii of gyration, and higher Cα densities. These findings imply that proteases are, as a group, more tightly packed than other proteins. There are also notable differences in secondary structure content between these two groups of proteins: proteases have fewer helices and more loops. We speculate that both high packing density and low α-helical content coevolved in proteases to avoid autolysis. By using the structural parameters that seem to show some separation between proteases and nonproteases, a neural network has been trained to predict protease function with over 86% accuracy. Moreover, it is possible to identify proteases whose folds were not represented during training. Similar structural analyses may be useful for identifying other classes of proteins and may be of great utility for categorizing the flood of structures soon to flow from structural genomics initiatives.
The genome sequencing projects currently underway have given birth to a new pursuit: determining the three-dimensional structures of an organism's proteome. This new endeavor, dubbed “structural genomics,” has an initial goal of solving the structures of proteins that have little or no sequence identity to proteins of known structure so as to map out protein fold space most efficiently and to provide modeling scaffolds for proteins of biomedical interest (1–3). Structures solved to meet this goal will include proteins of unknown function as has recently been reported (4, 5). Deducing the functions of proteins from their structures would be beneficial, because it could suggest possible roles for a much larger group of homologous proteins from other organisms.
Herein, we investigate whether a broad class of proteins of similar function, but not necessarily similar fold or catalytic mechanism, has distinguishing structural characteristics. We focus on the proteases, a very well studied class of proteins. Before the development of recombinant methods for protein expression, digestive enzymes were the subjects of many early structural and mechanistic studies, because they were easy to obtain in large quantities from natural sources (6). Today, the database of protease structures has grown to include a variety of molecules that play critical roles in many biological processes ranging from viral replication to the development and growth of an organism.
As with nearly all biological processes, protease activity must be regulated tightly. Regulation is particularly important for proteases, because all proteins, at some level, are their natural substrates. Different mechanisms have arisen for protease regulation. These include inhibition by specific protease inhibitors as well as synthesis as zymogens with covalently attached, inhibitory prosegments (7). Proteases may also be restricted to certain parts of the cell (e.g., the proteasome) or function only under specific environmental conditions (e.g., low pH).
Regardless of the mechanism of regulation, under conditions of optimal activity, all proteases must avoid inappropriate self-cleavage. Because the current structural database contains hundreds of protease structures from roughly a dozen evolutionarily unrelated families, we can ask whether common mechanisms for avoiding autolysis have coevolved in proteases as a whole.
Materials and Methods
Data Set Construction and Calculations.
The nonprotease data set for the statistical analysis was constructed from Hobohm and Sander's (8, 9) “pdb select” list of proteins with no more than 25% sequence identity and crystallographic resolution better than 2.5 Å. Only structures of biologically active, monomeric proteins were used. The structure selection criteria were the same for the proteases except that the sequence identity cutoff for proteases was 35% so as to include more examples. In both cases, the molecular mass range was 14–54 kDa. The final sets contained 36 protease and 154 nonprotease structures. The PDB identifiers of the proteases are listed in the text. The nonproteases were 153l, 16pk, 1a0p, 1a17, 1a26, 1a34, 1a6g, 1a8e, 1a9s, 1ad6, 1ads, 1ah7, 1ak0, 1ak1, 1ako, 1akz, 1alu, 1am7, 1amm, 1amx, 1anf, 1aoh, 1aol, 1aqb, 1arv, 1ash, 1asw, 1at0, 1atg, 1aua, 1auk, 1ax8, 1axn, 1azo, 1bc5, 1bd8, 1bg0, 1bgc, 1bgf, 1bjk, 1bkb, 1bol, 1bp1, 1brt, 1bv1, 1bxw, 1byb, 1byq, 1c25, 1c3d, 1ceo, 1cex, 1cfb, 1cfr, 1chd, 1ckn, 1cnv, 1cpo, 1csh, 1csn, 1dad, 1dhr, 1dhs, 1dhy, 1edg, 1fdr, 1fmt, 1fts, 1g3p, 1gky, 1gpl, 1grj, 1gso, 1ha1, 1hxn, 1idk, 1ihp, 1inp, 1ips, 1ixh, 1juk, 1lba, 1lcl, 1lit, 1lki, 1maz, 1mml, 1mpg, 1mrj, 1mrp, 1msk, 1mup, 1nar, 1nif, 1nkr, 1np4, 1npk, 1ois, 1opr, 1oyc, 1pda, 1pgs, 1phc, 1phm, 1pmi, 1pne, 1poc, 1pot, 1pta, 1pty, 1pud, 1qnf, 1qtq, 1ra9, 1rcf, 1rec, 1rhs, 1rss, 1rsy, 1sbp, 1tca, 1tde, 1tfr, 1thv, 1tib, 1tml, 1uae, 1uxy, 1v39, 1vhh, 1vid, 1vjs, 1wab, 1zin, 2abk, 2baa, 2cba, 2cyp, 2dri, 2end, 2gar, 2hft, 2i1b, 2liv, 2pia, 2plc, 2pth, 2sns, 2thi, 3nll, 3seb, 3sil, 4xis, and 6cel.
Surface areas were calculated by using the method of Lee and Richards (10) as implemented by the program calc-surface (11). A default probe radius of 1.4 Å was used, except when calculating surface roughness. In that case, radii of 1.25, 1.5, 1.75, and 2.0 Å were used. In-house utility programs were used to calculate radii of gyration, contact order (12), and interresidue Cα–Cα contacts. Cαs within 5.5 Å of one another were considered to be in close proximity. Contacts between residues at least three positions apart in sequence were considered. Secondary structure assignments were made by using the dssp program (13).
Neural Network Architecture.
The network used for prediction had a two-layer architecture with two hidden nodes in the first layer and a single output. Training was performed with a standard feed-forward, error back-propagation algorithm (14). Weights in the network were determined by using on-line learning with sum-of-squares error and a gradient-descent learning rule. A logistic activation function was used in all nodes.
The inputs used to train the neural network consisted of 10 data fields and a target value. The target value was set at 0.9 for true examples and at 0.1 for false examples. The input feature set included the percentage loop, the percentage helix, the percentage sheet, the average loop length, the average molecular mass per residue, the average number of contacts, the surface area per molecular mass, the contact order, the radius of gyration, and a zinc Boolean (1 if a structure contains Zn and 0 if it does not).
For the neural net testing, the nonprotease set was enlarged to 878 members. The examples in the false training and test sets were fixed, and no proteins in the false examples had greater than 45% sequence identity to any protein in the training set. Each of the original 36 proteases was tested by using a neural net that was trained on the remaining 35 proteases. For every experiment, the net was trained and tested five times, and the test results were averaged over all runs. For the prediction of protease classes, the protease example set was enlarged to 71 members with the highest homology between two members of different classes being 23.5% (resulting from one distantly related aminopeptidase/carboxypeptidase pair). Each class was removed individually and tested against the remaining proteases.
psi-blast.
The protease sequences used in the class experiment described above were used to develop sequence profiles to query the nonredundant sequence database with psi-blast (15); 10 iterations were performed on each of the proteases individually. An expect value of 1.0 was used for each iteration.
Results and Discussion
Two representative sets of protein structures (16, 17) were assembled, one containing 36 proteases and another containing 154 nonproteases of similar size. Proteases from 10 structural families were represented, including the trypsin-like serine proteases from three distantly related subfamilies (1dan, 1elt, 5gds, 1try, 1hne, 1cgh; 1exf, 1arb; 1sgp, 2alp), the subtilisin-like serine proteases (1gci, 2prk), cysteine proteases (1cjl, 1cv8, 3pbh, 8pch), aspartic proteinases (1bxo, 1eag, 1htr, 2asi), metzincins (1hfc, 1iab, 1iag, 1kuh, 1smp), thermolysin-like metalloproteases (1bqb, 1ezm, 1lml), aminopeptidases (1igb, 1lam, 1xjo), carboxypeptidases (1obr, 2ctc), and three proteases with unrelated folds (1ac5, 1lay, 1mat).
Fig. 1A shows surface area (10) plotted against molecular mass for the two sets of proteins, with a line fit to the nonprotease data points. Strikingly, 81% of the proteases fall below this line. These results are in accordance with a trend noted 8 years ago by one of us (18) with an independent set of 72 representative protein structures (19).
Figure 1.

(A) Molecular mass vs. surface area for proteases (♦) and nonproteases (▿). The linear fit for the nonproteases gives a correlation coefficient (R) of 0.95. The average surface area to molecular mass ratios for the proteases and nonproteases are 0.39 ± 0.03 Å2/Da and 0.43 ± 0.05 Å2/Da, respectively. (B) Radius of gyration versus molecular mass. R (nonproteases) = 0.96.
What physical attributes account for the smaller surface areas seen in proteases? Factors such as a smoother surface, tighter interatomic packing, or a more spherically symmetric shape could all contribute to the observed segregation of protease surface areas. The smoothness of protein surfaces may be calculated by determining the dependence of the surface area on the radius of the probe sphere used to map out the surface (20). The roughness or fractal dimension (D) of a surface is given by the relationship:
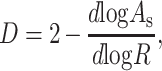 |
1 |
where R is the probe radius and As is the accessible surface area. A perfectly smooth surface will not depend on the probe size and will thus have a fractal dimension of two. Proteases and nonproteases were found to have very similar fractal dimensions (D = 2.17 in both cases). Thus, overt surface smoothness does not explain the smaller surface areas of proteases.
To assess how the shapes of proteases compare with those of nonproteases, the radius of gyration of each protein was computed (Fig. 1B). This measure is sensitive to the mass distribution of a protein. Proteases, particularly those smaller than 30 kDa, fall along the lower edge of the distribution, suggesting that they may be as compact and/or as spherically symmetrical as physically possible.
Residue packing was analyzed by computing the number of close contacts (≤ 5.5 Å) between Cα carbons. The proteases tend to have more contacts per residue than the nonproteases (Fig. 2), implying that the polypeptide backbone, on average, passes closer to itself in the proteases than in the nonproteases. The higher number of contacts per residue may be explained, in part, by proteases having slightly smaller amino acids on average. The average residue mass for the nonproteases in our data set is 111.2 ± 3.6 Da compared with 108.2 ± 4.8 Da for the proteases. Neither Cα contacts per residue nor surface area to molecular mass ratios are specific to a structural family; related family members (e.g., the various trypsin-like serine proteases) had widely scattered values for both of these measures.
Figure 2.

Number of Cα–Cα contacts within 5.5 Å of one another per residue for the nonprotease (░⃞) data set and the protease (■) data set. The average contacts per residue are 0.81 ± 0.08 for the proteases and 0.75 ± 0.07 for the nonproteases.
There is a striking difference between the secondary structure composition of the proteases and the nonproteases. Proteases have a significantly lower helical content: no protease has more than 43% of its residues in helices (Fig. 3A). Also, the fraction of residues in coil regions is significantly higher in the proteases than in the nonproteases (Fig. 3B). Proteases also tend to have longer loops (7.0 ± 1.8 residues as opposed to 6.1 ± 1.6 residues for the nonproteases).
Figure 3.

Secondary structure comparisons between the nonproteases (░⃞) and proteases (■). (A) Percentage helix. The averages are 24.1 ± 11.4 (proteases) and 36.0 ± 18.3 (nonproteases). (B) Percentage loop. The averages are 44.3 ± 5.1 (proteases) and 37.6 ± 8.0 (nonproteases). Categories are upper limits of bins.
Overall, it seems that proteases with diverse structures and catalytic mechanisms have subtle differences in several structural parameters from other proteins. The fact that these structural differences exist begs the question of whether they are due to the function of proteases. One obvious motive for lowering surface area, either through higher packing density or through a more spherically symmetric shape, would be to avoid autodegradation; i.e., to provide as few susceptible sites for cleavage as possible. Although substrate specificity may, to a large extent, prevent autolysis, at high local concentration, cleavage may be more difficult to avoid.
One might imagine that a protease of very broad specificity, of high activity, or with environmental exposure to other proteases would have the strongest need to avoid autolysis and thus would be packed most efficiently. Although there does not seem to be any correlation between specificity or activity and packing efficiency among the proteases in our data set, we do observe that proteases from unicellular organisms have lower surface area to molecular mass ratios and higher Cα densities than proteases from multicellular organisms. This observation agrees with Perona and Craik's (21) hypothesis that, in the chymotrypsin family of proteases, the higher order eukaryotic proteases tend to be more specialized. The authors speculate that organisms with specialized cell types require a diversity of protease functions and that their accompanying large genomes allow for protease gene duplication and subsequent specialization. Unicellular organisms, by contrast, have smaller, more efficient genomes and may produce only a small set of more general-purpose proteases. Organisms with smaller genomes and simpler lifestyles may also lack other means of regulating protease activity, such as specialized inhibitors, and may thus have to rely more on intrinsic means of resisting autolysis.
It is likely that the relatively low α-helical content of the proteases has also arisen as a mechanism to avoid autolysis. In general, proteolytic nick sites rarely occur in elements of secondary structure and are particularly rare in β-sheets (22, 23). Hubbard and coworkers (22, 24) have argued that an α-helix, being a local element of secondary structure, can unwind to accommodate a protease substrate binding site. Because a β-strand is already in an extended conformation and usually hydrogen bonded to one or two other strands, a more dramatic unfolding event would have to take place to expose the backbone of such a segment sufficiently.
We were surprised to discover that proteases have a higher than average loop content and longer than average loops. This result is an apparent contradiction to the antiautolysis argument made above. Evidence from both NMR and x-ray crystallographic studies of proteins indicates that loops are the most flexible regions of a protein. Furthermore, loops are usually targets in limited proteolysis experiments (24, 25) and make this technique useful for mapping protein topology and domain structure. However, as Hubbard and colleagues (26) have pointed out, not all loops are proteolytic targets. For example, loops that pack well against the protein surface will not be good candidates for proteolysis. Although there is no direct correlation between surface area to molecular mass or close Cα contacts/residue and the fraction of residues found in loop regions, the proteases are globally better packed as judged by both of these criteria. We suspect that the high loop content of proteases most likely reflects the bias against helical structure. As discussed above, β-structure has the advantage of making more hydrogen bond cross-links between sequentially distant residues and may serve to anchor down the chain so as to prevent unraveling and proteolysis. By traversing the extent of a globular protein by using extended strands, the polypeptide chain will need to make more reversals of direction. Such a protein will thus have a greater fraction of residues within loops. In fact, such a trend is evident in our data set of nonproteases. The predominantly β-proteins (those with less than 10% α-helical structure) contain 42 ± 7% of their residues in loops, whereas the predominantly α-proteins contain 30 ± 10% of their residues in loops. (The average sizes of the proteins in these sets are 231 residues for the α-proteins and 193 residues for the β-proteins.)
Although we cannot say for certain that the noted structural differences of proteases are related to function, we can nonetheless use these differences to classify structures as potential proteases. Given the overlapping distributions seen in Figs. 1–3 for the proteases and nonproteases, one structural difference by itself is clearly not sufficient to distinguish between them. However, when multiple properties are plotted simultaneously, the proteases are seen to cluster (Fig. 4), suggesting that it may be possible to predict protease function by using these and additional parameters.
Figure 4.

A combination of three metrics for the proteases (●) and nonproteases (○) shown in stereo.
In an attempt to recognize the cumulative significance of these structural differences, a neural network was trained by using the parameters that potentially show some discrimination between proteases and nonproteases. Each protease was, in turn, used as the sole true test example for a network trained on the remaining 35 proteases. In 31 of 36 cases (86%), the network was able to identify correctly the remaining protease as such. The mean performance on the 258 false examples tested was 87%, showing that the number of nonproteases erroneously called proteases is also small. Interestingly, several of the false positive structures were from the O-glycosidase family. Glycosidases may be subject to similar evolutionary constraints, because they may show cross-reactivity for peptide bonds.
A far more stringent test of the predictive power of the parameters we have identified would be to demonstrate that we can correctly identify novel structures as proteases. To address this issue, the network was retrained eight times, each time leaving out one family of proteases from training and then testing on this family (Table 1). The eight families have little sequence identity to each other, different folds, and different catalytic mechanisms. Therefore, this experiment simulates a prediction made on a novel structure. Although the prediction of two classes of proteases (aspartic and cysteine) tested poorly, the performance on the remaining classes was remarkably good. Most notably, when the largest subset of proteases, the trypsin-like serine proteases, is removed from the training set, the resulting network correctly identifies all of them as proteases.
Table 1.
Breakdown of neural net results when eight individual protease classes are tested
| Protease class | Proteases
|
Nonproteases
|
||
|---|---|---|---|---|
| Correct | Incorrect | Correct | Incorrect | |
| Aminopeptidases | 3 | 0 | 211 | 47 |
| Aspartic proteases | 4 | 9 | 212 | 46 |
| Carboxypeptidases | 4 | 0 | 207 | 51 |
| Cysteine proteases | 0 | 9 | 209 | 49 |
| Metzincins | 7 | 2 | 207 | 51 |
| Thermolysin-like metalloproteases | 5 | 0 | 208 | 50 |
| Subtilisin-like serine proteases | 8 | 0 | 207 | 51 |
| Trypsin-like serine proteases | 19 | 0 | 209 | 49 |
Each class is removed completely from the training set and then tested by using the neural network. Listed are the individual classes and the number of proteases and nonproteases the network assigned correctly and incorrectly.
The performance on the nonproteases in this experiment is slightly worse (80–82%) than in the training experiment that used all of the protease families simultaneously (87%). This result suggests that leaving out some information degrades performance on the false examples. In an actual structural genomics application, the network would be trained with all available information, even including protease structures of high sequence identity to one another. We would expect the performance on the nonproteases in this case to meet or exceed the 87% level.
Another measure of the success of this structure-based method is in comparison to sequence-based methods. We used psi-blast, an iterative sequence database search tool (15), to try and detect distant homologies between different protease classes. Only 3 of the 36 proteases had hits with an expectation value (the number of expected random hits given the query sequence and database size) lower than 1.0 between protease classes, and only 1 of these had an expectation value of less than 0.1. Thus, psi-blast is far less likely than our method to identify a novel protease structure. Although sequence-based methods are clearly powerful for detecting remote homologies resulting from divergent evolution, our results suggest that structure-based methods can detect convergent evolution. Both methods should probably be tested when classifying novel protein structures.
In summary, we have successfully trained a neural network to use global structural characteristics to predict protease function. We expect that similar approaches will work for other classes of proteins: our initial neural network predictive results with DNA-binding proteins have been encouraging. Along with the parameters already used, additional parameters, such as electrostatic charge distribution, predicted isoelectric point, and surface hydrophobicity, could be added to refine the prediction of other classes of proteins. It is also possible that some parameters, such as surface roughness, although not helpful in discriminating proteases from nonproteases, could be useful in distinguishing other classes. Methods such as the one described herein potentially could lead to the structural classification of a whole series of proteins and serve a primary role in structural genomics.
Acknowledgments
We thank John Diener and Yael Mandel-Gutfreund for helpful comments on the manuscript and Melissa Cline and David Haussler for invaluable discussions. This work was supported by National Institutes of Health Grant GM52885 and the University of California Biotechnology Research and Education Program.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.070548997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.070548997
References
- 1.Montelione G T, Anderson S. Nat Struct Biol. 1999;6:11–12. doi: 10.1038/4878. [DOI] [PubMed] [Google Scholar]
- 2.Kim S H. Nat Struct Biol. 1998;5,Suppl.:643–645. doi: 10.1038/1334. [DOI] [PubMed] [Google Scholar]
- 3.Terwilliger T C, Waldo G, Peat T S, Newman J M, Chu K, Berendzen J. Protein Sci. 1998;7:1851–1856. doi: 10.1002/pro.5560070901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hwang K H, Chung J H, Kim S, Han Y S, Cho Y. Nat Struct Biol. 1999;6:691–696. doi: 10.1038/10745. [DOI] [PubMed] [Google Scholar]
- 5.Zarembinski T I, Hung L W, Mueller-Dieckmann H J, Kim K K, Yokota H, Kim R, Kim S H. Proc Natl Acad Sci USA. 1998;95:15189–15193. doi: 10.1073/pnas.95.26.15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neurath H. Science. 1984;224:350–357. doi: 10.1126/science.6369538. [DOI] [PubMed] [Google Scholar]
- 7.Khan A R, James M N G. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hobohm U, Scharf M, Schneider R, Sander C. Protein Sci. 1992;1:409–417. doi: 10.1002/pro.5560010313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hobohm U, Sander C. Protein Sci. 1994;3:522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 11.Gerstein M. Acta Crystallogr A. 1992;48:271–276. [Google Scholar]
- 12.Plaxco K W, Simons K T, Baker D. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 13.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 14.Rumelhart D, McClellard J. Parallel Distributed Processing: Exploration in the Microstructure of Cognition. Cambridge, MA: MIT Press; 1986. [Google Scholar]
- 15.Altschul S F, Madden T L, Schaeffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abola E E, Sussman J L, Prilusky J, Manning N O. Methods Enzymol. 1997. 556–571. [DOI] [PubMed] [Google Scholar]
- 17.Sussman J L, Lin D, Jiang J, Manning N O, Prilusky J, Ritter O, Abola E E. Acta Crystallogr D. 1998;54:1078–1084. doi: 10.1107/s0907444998009378. [DOI] [PubMed] [Google Scholar]
- 18.Gregoret L M. Ph.D. Thesis. San Francisco: Univ. of California; 1991. [Google Scholar]
- 19.Gregoret L M, Cohen F E. J Mol Biol. 1990;211:959–974. doi: 10.1016/0022-2836(90)90086-2. [DOI] [PubMed] [Google Scholar]
- 20.Lewis M, Rees D C. Science. 1985;230:1163–1165. doi: 10.1126/science.4071040. [DOI] [PubMed] [Google Scholar]
- 21.Perona J J, Craik C S. J Biol Chem. 1997;272:29987–29990. doi: 10.1074/jbc.272.48.29987. [DOI] [PubMed] [Google Scholar]
- 22.Hubbard S J, Beynon R J, Thornton J M. Protein Eng. 1998;11:349–359. doi: 10.1093/protein/11.5.349. [DOI] [PubMed] [Google Scholar]
- 23.Fontana A, deLaureto P P, DeFilippis V, Scaramella E, Zambonin M. Folding Des. 1997;2:R17–R26. doi: 10.1016/S1359-0278(97)00010-2. [DOI] [PubMed] [Google Scholar]
- 24.Hubbard S J, Eisenmenger F, Thornton J M. Protein Sci. 1994;3:757–768. doi: 10.1002/pro.5560030505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fontana A, Fassina G, Vita C, Dalzoppo D, Zamai M, Zambonin M. Biochemistry. 1986;25:1847–1851. doi: 10.1021/bi00356a001. [DOI] [PubMed] [Google Scholar]
- 26.Hubbard S J. Biochim Biophys Acta. 1998;1382:191–206. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]