

Abstract
Originally identified as axon guidance molecules, semaphorins are now known to be widely expressed mediators that play significant roles in immune responses and organ morphogenesis. However, not much is known about the signaling pathways via which they exert their organ-specific effects. Here we demonstrate that Sema4A, previously identified as an activator of T-cell-mediated immunity, is expressed in endothelial cells, where it suppresses vascular endothelial growth factor (VEGF)-mediated endothelial cell migration and proliferation in vitro and angiogenesis in vivo. Mice lacking Sema4A exhibit enhanced angiogenesis in response to VEGF or inflammatory stimuli. In addition, binding and functional experiments revealed Plexin-D1 to be a receptor for Sema4A on endothelial cells, indicating that Sema4A exerts organ-specific activities via different receptor-mediated signaling pathways: via Plexin-D1 in the endothelial cells and via T-cell immunoglobulin and mucin domain-2 in T cells. The effects of Sema4A on endothelial cells are dependent on its ability to suppress VEGF-mediated Rac activation and integrin-dependent cell adhesion. It thus appears that Sema4A–Plexin-D1 signaling negatively regulates angiogenesis.
Keywords: angiogenesis, Plexin-D1, Semaphorin 4A, VEGF
Introduction
The development of the vertebrate vasculature involves the differentiation and proliferation of endothelial precursor cells and their coalescence into the tubes that comprise the primary vascular plexus, a process called vasculogenesis. Subsequent remodeling of the primary vascular plexus into a highly branched hierarchical vascular tree composed of arteries and veins involves a process called angiogenesis. Although a variety of vasculogenic and angiogenic factors (e.g., vascular endothelial growth factor (VEGF) and angiopoietin-1) involved in vascular endothelial cell specification and differentiation have been identified; it remains unclear how the stereotypic patterning of newly formed vessels is determined, although several lines of evidence suggest that axon guidance molecules such as semaphorin, Netrin, Epherin and Slit play key roles in vascular path finding and network formation (Carmeliet and Tessier-Lavigne, 2005; Eichmann et al, 2005).
Characterized by a conserved sema domain, semaphorins are a family of membrane-bound and secreted factors that function as potent axonal repellants in the nervous system (Kolodkin et al, 1993; Luo et al, 1993). In addition, prototypical class 3 semaphorins have been shown to be involved in vascular development. For instance, Sema3A is expressed on endothelial cells within developing vessels and inhibits endothelial cell migration by interfering with integrin function (Serini et al, 2003). In addition, Sema3E repels developing intersomitic vessels (Gu et al, 2005), but its N-terminal region (61 kDa) promotes migration of cells from an endothelial cell line (Christensen et al, 2005) and Sema3F inhibits tumor angiogenesis and metastasis in a mouse model (Bielenberg et al, 2004; Kessler et al, 2004). Class 3 semaphorins such as Sema3A and Sema3F directly bind to Neuropilin family proteins (Kolodkin et al, 1997) and transduce signals via a co-receptor, type A Plexin. In addition to heterodimer formation with type A Plexin, Neuropilin-1 binds to VEGF165 (Soker et al, 1998) and forms a complex with VEGFR2 on endothelial cells, thereby enhancing downstream VEGF signaling (Miao et al, 1999). In fact, mice lacking Neuropilin-1 exhibit defects in embryonic vascular development (Kawasaki et al, 1999), suggesting Sema3A and Sema3F influence vessel formation via Neuropilin-1. On the other hand, Neuropilin knock-in mice that express an altered form of Neuropilin-1 capable of binding VEGF165 but not semaphorins exhibit a severely affected nervous system, but normal vascular development (Gu et al, 2003), indicating that Semaphorin–Neuropilin-1 signaling is dispensable for vascular development. Notably, Sema3E binds directly to Plexin-D1 (Gu et al, 2005) so that mice lacking Plexin-D1 or Sema3E exhibit similar defects in intersomitic vessel patterning (Gitler et al, 2004; Torres-Vazquez et al, 2004; Gu et al, 2005). That said, the molecular mechanisms by which class 3 semaphorins regulate vessel formation remains to be fully explained.
In the present study, we found that Sema4A, the membrane-bound class 4 semaphorin expressed on endothelial cells, directly binds to Plexin-D1 and negatively regulates VEGF-dependent vascular formation. Sema4A was previously found to exert effects on immune system function by inducing T-cell activation via Tim-2, a member of the T-cell immunoglobulin and mucin domain (Tim) protein family expressed on the surface of T cells (Kumanogoh et al, 2002). Thus, Sema4A appears to exert distinct biological effects via separate receptor-mediated signaling pathways.
Results
Anti-angiogenic activity of Sema4A in endothelial cells
Whole mount in situ hybridization showed that Sema4A mRNA is expressed specifically within intersomitic blood vessels (Figure 1A–C), an expression pattern similar to that of Plexin-D1 (Figure 1D–F) in developing mouse embryos. This suggests that Sema4A plays a functional role in vascular formation, which prompted us to evaluate its in vitro effects on human umbilical vein endothelial cells (HUVEC). We initially used a Transwell system to examine Sema4A's ability to stimulate endothelial cell migration. After coating both sides of the Transwell filters with fibronectin to allow suitable integrin engagement, VEGF165 was added to the lower chambers, alone or in combination with soluble Fc-fused semaphorins (Supplementary Figure 1). The migration of HUVECs, which was increased two-fold by VEGF165 alone, was suppressed by Sema4A-Fc (Figure 1G). VEGF165-activated migration of HUVECs was also suppressed by Sema3E-Fc, but was somewhat promoted by Sema4D-Fc.
Figure 1.
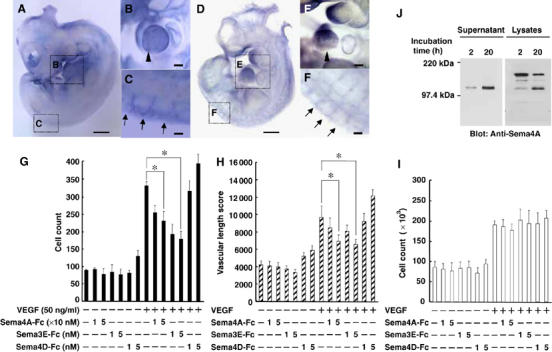
Sema4A suppresses VEGF165-mediated angiogenesis. (A) Whole-mount in situ hybridization of an E10.5 mouse embryo shows Sema4A expression. The magnified views of the embryo reveal Sema4A mRNA expression within the cardiac ventricle (B, arrowhead) and the dorsal ends of intersomitic blood vessels (C, arrows). (D) Whole-mount in situ hybridization of an E10.5 mouse embryo reveals Plexin-D1 expression. The magnified views of the embryo reveal Plexin-D1 mRNA expression within the cardiac ventricle (E, arrowhead) and the intersomitic blood vessels (F, arrows). Scale bars, 100 μm for (A) and (D), and 20 μm for (B), (C), (E) and (F). (G–I) Effect of dose-dependent suppression of Sema4A-Fc on VEGF165-mediated HUVEC migration (G), in vitro angiogenesis (H) and proliferation (I). In the absence of VEGF165, Sema4A-Fc, Sema3E-Fc and Sema4D-Fc did not induce significant angiogenic responses. VEGF165 (50 ng/ml) induced significant angiogenic responses that were suppressed by Sema4A-Fc (10–50 nM) and Sema3E-Fc (1–5 nM), but not by Sema4D-Fc (1–5 nM). Bars represent the means±s.e.m. of 4–7 experiments for each condition. *P<0.05 versus the value in the presence of VEGF165 without Sema4A-Fc, Sema3E-Fc or Sema4D-Fc. (J) Release of a soluble form of Sema4A from the cell surface. HEK293 cells expressing Sema4A were surface-labeled with biotin and then cultured for the indicated times. Cell lysates (1% NP-40) or culture supernatants were immunoprecipitated with anti-Sema4A antibody, after which the immunoprecipitates were subjected to SDS–PAGE and probed with streptavidin–POD conjugate.
Angiogenic factors such as VEGF stimulate endothelial cells to form capillary-like tubular structures in collagen gel. Called in vitro angiogenesis, this process correlates well with the ability of endothelial cells to form blood vessel in vivo in response to the same stimuli (Korff and Augustin, 1998; Conrotto et al, 2005). We then carried out an in vitro angiogenesis assay in which endothelial cells were co-cultured with fibroblasts in a collagen gel in the presence or absence of semaphorins (Figure 1H). After 11 days, the biological response was evaluated in terms of total length of the tubular structure per area (Supplementary Figure 2). We found that Sema4A-Fc and Sema3E-Fc, but not Sema4D-Fc, suppressed VEGF165-induced formation of tubular structures composed of HUVECs. The optimal biological concentration of Sema4A-Fc (10–50 nM) was 10 times higher than that of Sema3E-Fc (1–5 nM), which is comparable to the optimal concentration of Sema4D and VEGF165 (3.5 and 2 nM, respectively) (Conrotto et al, 2005).
VEGF is also known to stimulate endothelial cell proliferation (Yancopoulos et al, 2000; Zachary, 2003). Consistent with this finding, VEGF165 elicited a two-fold increase in the proliferation of HUVECs, which was unaffected by Sema4A-Fc, Sema3E-Fc or Sema4D-Fc (Figure 1I).
The results summarized above demonstrate that a recombinant soluble form of Sema4A suppresses migration of endothelial cells and their organization into capillary-like structures. This suggests that membrane-bound Sema4A may exert its activity after conversion into a soluble form. We tested this idea in surface biotinylation-chase experiments, in which the fate of the biotinylated Sema4A on the cell surface was observed over a period of 20 h (Figure 1J). SDS–PAGE and Western blot analysis revealed that a single 120-kDa band was immunoprecipitated from the culture supernatants of cells expressing full-length Sema4A, and its intensity increased with longer culture times. This suggests that a soluble form of Sema4A is cleaved from membrane-bound Sema4A. Consistent with this idea, two species of Sema4A with molecular weights of about 150 and 120 kDa were observed in lysates of cells expressing full-length Sema4A. Moreover, the intensity of the 150-kDa band, which corresponded to membrane-bound Sema4A, declined as the 120-kDa band emerged. It is thus plausible that Sema4A exerts its activity as locally released protein.
Anti-angiogenic activity of Sema4A in vivo
We then explored the in vivo effects of Sema4A on embryonic vascularization. Normally, chick embryos (HH stage 4) initially form primitive tubular networks of proliferating endothelial cells, which are then remodeled through both pruning and vessel enlargement to form the interconnecting branching pattern characteristic of the mature vasculature in embryos treated with control solution (Figure 2A–C) and embryos treated with control human IgG (Figure 2D–F). When chick embryos were incubated in the presence of Sema4A-Fc or Sema3E-Fc, however, they developed only a honeycomb-like primitive vascular plexus with no major vessels (Figure 2G–L).
Figure 2.
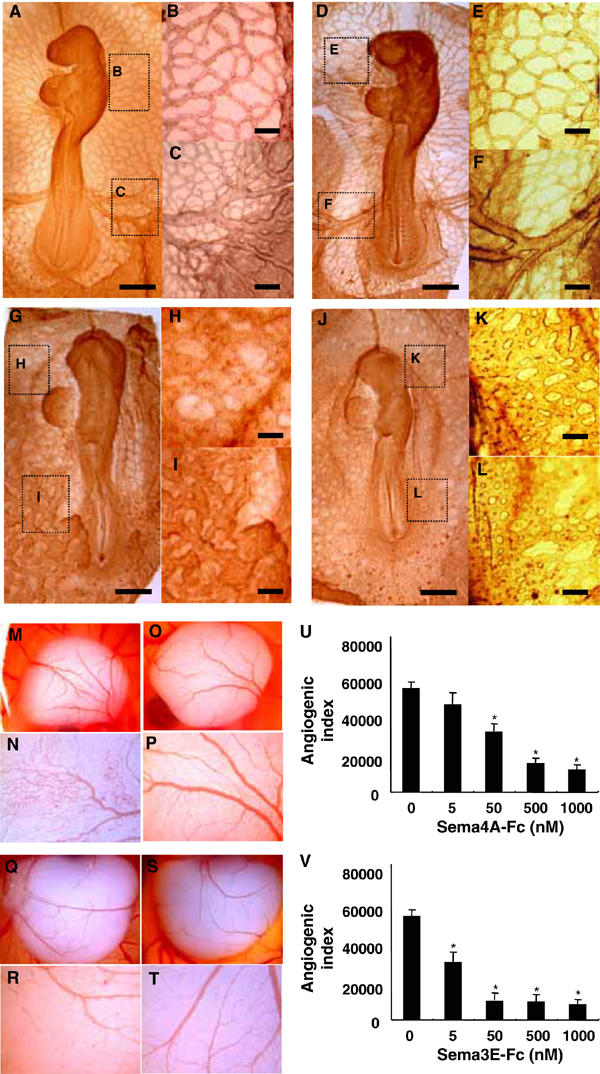
Sema4A suppresses angiogenesis during development. (A–L) Chick embryos on collagen gel cultures were exposed to control solution (A–C), control human IgG (50 nM) (D–F), Sema4A-Fc (50 nM) (G–I) or Sema3E-Fc (5 nM) (J–L) for 24 h. The magnified views of the embryo show a microvascular network (B, E, H, K) and major vessels (C, F, I, L). Sema4A-Fc and Sema3E-Fc suppress the formation of major vessels interconnecting with the microvascular network, which is characteristic of mature vasculature. Scale bars, 100 μm for A, D, G and J and 20 μm for B, C, E, F, H, I, K and L. (M–V) Chicken CAMs were implanted with sponges soaked with control buffer (M, N: n=10), control human IgG (O, P: n=10), Sema4A-Fc (50 nM) (Q, R: n=20) or Sema3E-Fc (5 nM) (S, T: n=20) and incubated for 3days. The magnified views of the embryo (N, P, R, T) show that Sema4A-Fc and Sema3E-Fc suppress vascular sprouting from vessels into previously avascular areas of CAMs. (U) Dose-dependent suppression of angiogenic indexes by Sema4A-Fc or Sema3E-Fc was assessed by estimating newly formed microvessels within previously avascular areas of CAMs. The bars represent means±s.e.m. of 10 CAM assays for each condition. *P<0.05 versus. the value of CAM exposed without Fc-fused proteins.
To assess further the role played by Sema4A in embryonic vessel formation, we carried out chorioallantoic membrane (CAM) assays, which is an in vivo model widely used to study neoangiogenesis. Gelatin sponges soaked in control solution, control human IgG, Sema4A-Fc or Sema3E-Fc were inserted into 8-day chicken embryo CAMs, which were then evaluated with respect to the appearance of newly formed blood vessels after a 3-day incubation. We found that whereas a normal vasculature was detectable around the sponges containing control solution (Figure 2M and N) or control human IgG (Figure 2O and P), sponges containing purified Sema4A-Fc or Sema3E-Fc were surrounded by poorly formed allantoic vessels (Figure 2Q–T), which is consistent with the apparent anti-angiogenic properties of Sema4A and Sema3E. Indeed, the angiogenic indexes were dose-dependently reduced in response to Sema4A-Fc or Sema3E-Fc, such that 10 times more Sema4A-Fc was required to exert the suppressive effect exerted by Sema3E-Fc (Figure 2U).
We analyzed the in vivo role played by Sema4A during vascular development using Sema4A-deficient mice. Macroscopic and histological examination of the embryos and adult mice revealed no apparent defects in the systemic vasculature (data not shown) (Kumanogoh et al, 2005), indicating that Sema4A is dispensable for embryonic vessel formation. We then examined the neoangiogenic response of the avascular cornea to an inflammatory stimulus (suture), and found that neovessel formation became evident 2–3 days after suturing the cornea and gradually progressed until after about 12 days it involved the entire cornea (Figure 3A). Three days after suturing, more extensive corneal neovascularization was seen in Sema4A-deficient mice than in wild-type mice, but by day 8 the levels of neovascular development were similar in the two mouse types (Figure 3B).
Figure 3.
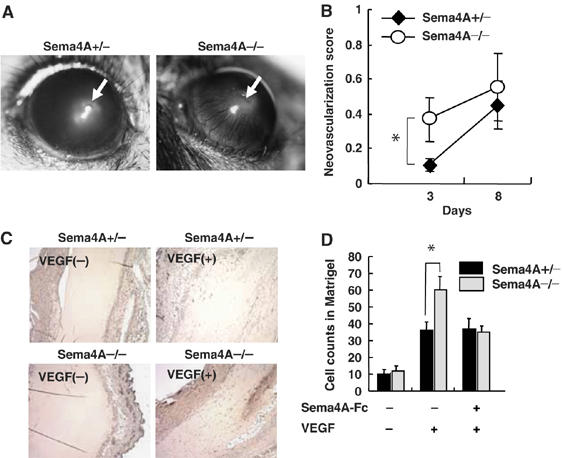
Knockdown of Sema4A promotes angiogenesis in vivo. (A) Transcorneal silk suture induces inflammation-associated neovascularization. Newly formed vessels growing outward from the suture were estimated 3 and 8 days after placement. (B) Quantification of newly formed vessels in the cornea shows greater vessel formation in Sema4A-deficient mice than in wild-type mice on day 3. The points represent the means±s.e.m. of six mice for each condition. *P<0.05 versus the value obtained with heterozygous mice implanted with suture. (C) Matrigel plugs containing VEGF165 with or without Sema4A-Fc were implanted in wild-type and Sema4A-deficient mice. Sections of Matrigel plugs excised 7 days after implantation were immunostained with anti-CD31 antibody. (D) Microvessels labeled with anti-CD31 antibody were counted in fields of equal area in sections of Matrigel plugs. The bars represent the means±s.e.m. of six mice for each condition. *P<0.05 versus the value of heterozygous mice implanted with corresponding Matrigel plugs.
We also used Matrigel plug assays to assess growth factor-induced angiogenesis in wild-type and Sema4A-deficient mice. We found that VEGF165 induced growth of neovessels into the Matrigel plugs in both wild-type and Sema4A-deficient mice (Figure 3C and D), but the effect was more pronounced in the latter. Moreover, the enhanced neovessel formation seen in Sema4A-deficient mice could be reversed by including Sema4A-Fc in the Matrigel plug (Figure 3D). Thus, neovascularization in response to angiogenic factors such as VEGF and inflammatory stimuli appears to be negatively regulated by Sema4A in adult mice.
Plexin-D1 is a receptor for Sema4A in endothelial cells
To investigate the molecular mechanism responsible for the observed biological effects of Sema4A, we initially studied whether Sema4A affects the binding of VEGF165 to its receptor, VEGFR2. By crosslinking experiment using 125I-labeled VEGF165, crosslinked complexes of 125I- labeledVEGF165 to VEGFR2 and Neuropilin-1 on HUVECs were identified on the basis of their molecular weights described previously (Soker et al, 2002; Figure 4A). The densities of these bands were not affected by the excess amounts of Sema4A-Fc (Figure 4A and Supplementary Figure 3), indicating that Sema4A does not compete with VEGF165 for VEGFR2 on endothelial cells. We then searched for candidate receptors for Sema4A (Figure 4B) among the various Neuropilins and Plexins expressed by endothelial cells (Soker et al, 1998; Miao et al, 1999; Basile et al, 2004; Gitler et al, 2004; Torres-Vazquez et al, 2004). Tim-2, a Sema4A receptor expressed in the immune system (Kumanogoh et al, 2002), was not detected on endothelial cells (data not shown). When we evaluated 125I-labeled Sema4-Fc binding to the membranes of cells expressing Plexin family proteins (Figure 4B), we detected binding to those expressing one of the Plexin-B proteins or Plexin-D1, but not to cells expressing Plexin-A proteins (Figure 4B, upper panel, arrowhead), Neuropilins or VEGFR2 (data not shown). To assess further the involvement of Plexin-B proteins and Plexin-D1, which are endogenously expressed in HUVECs (Figure 4C), in Sema4A-mediated endothelial cell activity, we examined the effect of Sema4A-Fc on the migration of HUVECs treated with siRNAs specific for the indicated Plexin family members (Figure 4D). Each siRNA strongly and exclusively suppressed expression of a Plexin-B family member or Plexin-D1 (Supplementary Figure 4), but only knockdown of Plexin-D1 expression interfered with the suppressive effect of Sema4A on VEGF-mediated cell migration, indicating that it is Plexin-D1 that serves as the Sema4A receptor in this case.
Figure 4.
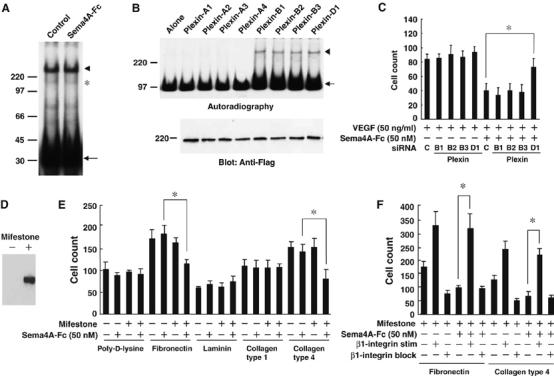
Sema4A binds to Plexin-D1 and suppresses integrin-mediated cell-matrix adhesion. (A) Crosslinking of 125I- VEGF165 (50 ng/ml) to its receptors on HUVECs in the presence and absence of Sema4A-Fc (50 nM). Autoradiography shows crosslinked complexes between 125I-VEGF165 and its receptors (VEGFR (arrowhead) and Neuropilin (asterisk)) are unaffected by the presence of excess amounts of unlabeled Sema4A-Fc. The uncoupled 125I- VEGF165 is indicated by the arrow. (B) Crosslinking of 125I-Sema4A-Fc (50 nM) to Plexin family members. Upper panel: autoradiography shows crosslinked complexes between 125I-Sema4A-Fc and the receptors (arrowhead) and uncoupled 125I-Sema4A-Fc (arrow). Lower panel: Immunoblot of cell lysates from transfected cells shows that amounts of receptors are almost identical. (C) Migration of HUVECs treated with the indicated siRNA. VEGF165-mediated migration of HUVECs is suppressed by Sema4A-Fc. This suppressive effect is reversed by treatment with siRNA specific for Plexin-D1, but not by any other siRNAs. The bars represent the means±s.e.m. for each condition. *P<0.05 versus the counts of migrating HUVECs treated with control siRNA in the presence of VEGF165 and Sema4A-Fc. (D) Mifestone-dependent induction of Plexin-D1 expression in HEK293 cells stably transfected with pGene/V5-His vector containing Plexin-D1 cDNA. (E, F) Adhesion of cells containing a Plexin-D1 gene-switch system to various matrices in the presence of VEGF165 (50 ng/ml). (E) Induction of Plexin-D1 by mifestone induces a suppressive effect of Sema4A-Fc on cell adhesion to fibronectin and collagen type IV. *P<0.05 versus cells treated with Sema4A-Fc alone. (F) The suppressive effect of Sema4A-Fc on adhesion of cells expressing Plexin-D1 to matrices is inhibited by HUST-4, a β1-integrin stimulatory antibody (β1-integrin stim), but not by JBS5, a β1-integrin blocking antibody (β1-integrin block). *P<0.05 between cells treated with Sema4A-Fc alone and cells treated with Sema4A-Fc in the presence of β1-integrin stimulatory antibody.
It was previously demonstrated that proper integrin function is required for angiogenesis (Hynes, 2002; Serini et al, 2006) and that class 3 semaphorin (Sema3A) regulates vascular formation by inhibiting integrin function (Serini et al, 2003). To determine whether Sema4A regulates integrin function through Plexin-D1, we established cell lines containing a gene-switch system in which Plexin-D1 expression was induced by mifestone (Figure 4D). Using this system, we found that Sema4A-Fc reduced cell adhesion to fibronectin and collagen type IV only after induction of Plexin-D1 expression by mifestone (Figure 4E). Thus, the interactions between fibronectin and its receptor, α5β1 integrin, and between collagen IV and its receptor, α1β1 integrin, were suppressed by Sema4A-Plexin-D1 signaling. Consistent with this finding, we also observed that Sema4A's suppressive effects were cancelled by a β1-integrin stimulating antibody (Figure 4F).
We finally measured the affinity with which Sema4A binds to Plexin-D1 using surface plasmon resonance assays in which the Flag-fused soluble recombinant extracellular domain of Plexin-D1 (Flag Plexin-D1-EC) was immobilized on the surface of a Biacore sensor chip via an anti-Flag antibody (Figure 5). By injecting increasing concentrations of Sema4A-Fc proteins over the sensor surface containing the immobilized Plexin-D1-EC, the typical profile of the protein–protein interaction was observed. At equilibrium, the binding response was calculated to have a KD value of 8.7 × 10−9 M (Figure 5A). For comparison, the KD value for the binding of Sema3E-Fc to Plexin-D1 was measured to be 1.3 × 10−9 M, that is, Sema3E-Fc bound to Plexin-D1 with about seven times the affinity of Sema4A-Fc (Figure 5B). The KD value for Sema3E-Fc to Plexin-D1 estimated by Biacore appeared to be 10 times lower than that estimated by Scatchard analysis using alkaline phosphatase (AP)-Sema3E (Gu et al, 2005), probably because Sema3E-Fc lacks the Ig domain, which may stabilize the binding of class 3 semaphorin to Neuropilin (Chen et al, 1998).
Figure 5.
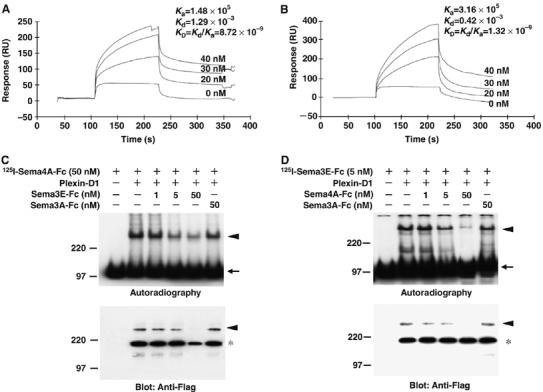
Sema4A directly binds to Plexin-D1 via its extracellular region. (A, B) BIAcore analysis of the interaction of Sema4A-Fc and Plexin-D1 (A) or Sema3E-Fc and Plexin-D1 (B). The indicated concentrations of Sema4A-Fc or Sema3E-Fc were sequentially injected through a flow cell within which recombinant Plexin-D1-FLAG was immobilized via anti-FLAG antibody on the sensor chip. The binding of Sema4A-Fc to Plexin-D1 and Sema3E-Fc to Plexin-D1 show an affinity constant (KD) of 8.72 × 10−9 and 1.32 × 10−9, respectively. (C) The binding competition experiment using 125I-Sema4A-Fc (50 nM) to Plexin-D1 in the presence of various amounts of unlabeled Sema3E-Fc (1–50 nM) or Sema3A-Fc (50 nM). Upper panel: Autoradiography shows 125I-Sema4A-Fc crosslinked complexes (300 kDa; arrowhead) and uncoupled 125I-Sema4A-Fc (100 kDa; arrow). Lower panel: Immunoblot shows the crosslinked Plexin-D1 (300 kDa; arrowhead) and the uncrossed Plexin-D1 (200 kDa; asterisk). Note that density of crosslinked complexes (300 kDa) decreases in parallel with the increasing amounts of unlabeled Sema3E-Fc but not Sema3A-Fc. (D) The binding competition experiment using 125I-Sema3E-Fc (5 nM) to Plexin-D1 in the presence of various amounts of unlabeled Sema4A-Fc (1–50 nM) or Sema3A-Fc (50 nM). Upper panel: Autoradiography shows 125I-Sema3E-Fc crosslinked complexes (300 kDa; arrowhead) and uncoupled 125I-Sema3E-Fc (100 kDa; arrow). Lower panel: Immunoblot shows the crosslinked Plexin-D1 (300 kDa; arrowhead) and the uncrossed Plexin-D1 (200 kDa; asterisk). Note that density of crosslinked complexes (300 kDa) decreases in parallel with the increasing amounts of unlabeled Sema4A-Fc but not Sema3A-Fc.
We then carried out competitive binding experiments to test whether Sema4A-Fc and Sema3E-Fc bind to overlapping sites on Plexin-D1. We found that 125I-labeled Sema4A-Fc binding to membranes of cells expressing Plexin-D1 was suppressed by excess Sema3E-Fc; conversely, 125I-labeled Sema3E-Fc binding to the membranes of cells expressing Plexin-D1 was suppressed by excess amount of Sema4A-Fc (Figure 5C and D). Thus, Sema4A and Sema3E bind to Plexin-D1 at overlapping site.
Sema4A inhibits downstream VEGF signaling
Consistent with our finding that Sema4A-Fc did not compete with VEGF165 for binding to VEGFR2, tyrosine phosphorylation of VEGFR2, which is the initial step in VEGF signal transduction, was unaffected by Sema4A-Fc (Figure 6A). Instead, VEGF-mediated activation of Rac, a regulator of cytoskeletal remodeling and cell adhesion (Burridge and Wennerberg, 2004), was significantly suppressed by Sema4A-Fc (Figure 6B). In parallel with the VEGF-mediated Rac activation, tyrosine phosphorylation of VEGFR2 is followed by the phosphorylation of Akt and Erk-1/2, which contribute to the survival and proliferation of endothelial cells, respectively (Zachary, 2003). Sema4A-Fc inhibited the phosphorylation of Akt, but only affected the phosphorylation of Erk-1/2 to a small degree (Figure 6C and D). Sema4A thus appears to inhibit VEGF-induced Rac and Akt activation, which in turn affects vascular migration and cell survival, respectively.
Figure 6.

Sema4A suppresses VEGF165-mediated signaling. (A–D) HUVECs exposed for 5 h to control medium or Sema4A-Fc (50 nM) were further cultured in the absence or presence for VEGF165 (50 ng/ml) for the indicated times, after which they were lysed for immunoprecipitation (IP) and/or immunoblot (Blot) assays. (A) Time course of tyrosine phosphorylation of VEGFR2 by VEGF165. The tyrosine-phosphorylated VEGFR2 was immunoprecipitated using anti-VEGFR2 antibody and detected using anti-phospho-tyrosine antibody (4G10). To ensure comparable loading, total VEGFR2 protein was immunoblotted with anti-VEGFR2 antibody. (B) Time course of Rac activation by VEGF165. Rac1-GTP was measured in pull-down assays using the GST-fused CRIB domain of PAK. Total Rac1 protein was immunoblotted with anti-Rac1 antibody. (C) Time course of the phosphorylation of Akt by VEGF165. Phosphorylated Akt and total Akt were immunoblotted with anti-phospho-Akt antibody and anti-Akt antibody, respectively. (D) Time course of the phosphorylation of Erk by VEGF165. Phosphorylated Erk and total Erk were immunoblotted with anti-phospho-Erk antibody and anti-Erk antibody, respectively. (E) Micrographs of HUVECs in response to VEGF165 (50 ng/ml) and Sema4A-Fc (50 nM). HUVECs coinfected with or without adenoviruses containing mutant Rac and GFP were cultured in the indicated reagent for 5 h, and then stained with phalloidin–rhodamine. HUVECs expressing mutant Rac were identified by the coexpression of GFP. VEGF-mediated protrusion of lamellipodia (arrowhead) was suppressed by Sema4A-Fc (asterisk) and by treatment with RacN17 (star). The suppressive effect of Sema4A-Fc was cancelled by treatment with RacV12 (arrow). (F) Effects of mutant Rac on VEGF-mediated HUVEC migration. Twenty-four hours after coinfection with adenoviruses containing mutant Rac and GFP, cells were used for migration assay in the presence of VEGF165 (50 ng/ml) with or without Sema4A-Fc (50 nM). HUVECs expressing mutant Rac were identified by the coexpression of GFP. VEGF-mediated migration of GFP-positive HUVECs was suppressed by Sema4A-Fc and by RacN17. The suppressive effect of Sema4A-Fc was cancelled by treatment with RacV12. *P<0.05 versus cells treated with Sema4A-Fc in the presence of VEGF165.
Given the recent finding that VEGF regulates cell-matrix adhesion during vessel formation (Avraham et al, 2003; Nikolopoulos et al, 2004), we evaluated evoked cytoskeletal remodeling in endothelial cells by staining actin fibers with Texas-Red phalloidin (Figure 6E and Supplementary Figure 5). We found that VEGF165 induced formation of actin stress fibers and protrusion of lamellipodia in HUVECs (Figure 6E, arrowhead). To determine whether Rac is required for the cytoskeletal rearrangement mediated by VEGF and Sema4A, we examined the morphological effects of VEGF165 and/or Sema4A-Fc on HUVECs overexpressing constitutively active Rac (RacV12) or dominant-negative Rac (RacN17). The VEGF-mediated protrusion of lamellipodia was suppressed by pretreatment with Sema4A-Fc in untransfected HUVECs (asterisk), but not in HUVECs transfected with RacV12 (Figure 6E, arrow), whereas VEGF-mediated lamellipodia was suppressed in HUVECs transfected with RacN17 (Figure 6E, star). Consistent with this finding, we observed that the suppressive effect of Sema4A-Fc on VEGF-mediated migration of HUVECs was cancelled by transfection with RacV12 and that VEGF-mediated migration of HUVECs was suppressed by transfection with RacN17 (Figure 6F). Taken together, these results suggest that Sema4A-Fc suppresses VEGF-mediated angiogenesis by downregulating Rac-GTP-dependent cytoskeletal rearrangement.
Discussion
We have shown that Sema4A is endowed with anti-angiogenic properties similar to those of known endogenous anti-angiogenic molecules, including endostatin, thrombospondin and tumstatin (Sund et al, 2005). Our findings indicate that Sema4A suppresses endothelial cell migration, a key component of all of the steps of vascular morphogenesis (Yancopoulos et al, 2000; Zachary, 2003), as well as VEGF-induced formation of well-organized tubular structures from endothelial cells, events that occur during angiogenesis in vivo. These in vitro results were strongly supported by in vivo assays making use of the chick CAM and in Matrigel angiogenesis assays carried out in mice lacking Sema4A. We then used two different experimental approaches to identify Plexin-D1 as a receptor for Sema4A on endothelial cells (an in vitro binding assay and a functional assay) and showed that Sema4A binds to Plexin-D1, which mediates its antiangiogenic activity. Within the immune system, Sema4A binds to Tim-2 on T cells, thereby activating their proliferation and differentiation (Kumanogoh et al, 2002, 2005). However, Sema4A actually shows greater affinity for Plexin-D1 (Kd=8.7 × 10−9) than for Tim-2 (Kd=7.0 × 10−8), suggesting that Sema4A's primary function under physiological conditions is to inhibit angiogenesis.
It is noteworthy that Sema3E is also a ligand for Plexin-D1 (Gu et al, 2005). We and Gu et al (2005) demonstrated that Sema3E acts via Plexin-D1 to serve as a chemorepellant of endothelial cells. Nevertheless, Sema4A and Sema3E do not have completely overlapping effects, whereas Sema3E released from somites guides intersomitic vessels into the corridor between somites (Gu et al, 2005), Sema4A does not, despite its expression in intersomitic vessels. Moreover, whereas Sema3E is a soluble ligand that exerts its effects over a fairly extensive area, Sema4A exerts its effects in a more localized manner, acting either as a membrane-bound or locally released protein. In addition, Plexin-D1 activation by the two ligands differs in terms of the kinetics, as the affinity for Plexin-D1 of Sema4A-Fc is seven times lower than that of Sema3E-Fc, which is also consistent with Sema4A exerting its effects in a more spatially restricted manner. Alternatively, it may be that the activities of the recombinant Fc-fused proteins used in this study do not fully reflect their native roles in endothelial cell biology. It has been demonstrated that the chemorepellant activity of class 3 semaphorins is associated with the dimeric forms of full-size molecules and that furin-dependent processing downregulates this activity (Adams et al, 1997; Klostermann et al, 1998; Koppel and Raper, 1998). In particular, the processing converts Sema3E from a chemorepellant into a chemoattractant in metastatic cancer cells and endothelial cells (Christensen et al, 2005), although the Sema3E-Fc used in this study tends to form a dimer rather than a monomer (Supplementary Figure 1). Thus, the functions of Sema4A and Sema3E in vivo should be determined by tissue-specific characteristics, including the capacity to alter the structures of the semaphorins.
Our use of genetically manipulated mice enabled us to obtain additional details of the anti-angiogenic activity of Sema4A under physiological conditions. Mice lacking Sema4A did not exhibit hypervascularity during development, but exhibited enhanced neoangiogenesis early during wound healing. Given that mice lacking Plexin-D1 exhibit severely defective angiogenic activity within cardiovascular system (Gitler et al, 2004; Torres-Vazquez et al, 2004), it seems plausible that one or more ligands other than Sema4A or Sema3E exert angiogenic effects via Plexin-D1. Consistent with that idea, the enhanced neoangiogenesis observed in mice lacking Sema4A after VEGF165 or injury was only transient. Moreover, mice lacking Sema3E, which exhibit a disrupted intersomitic vasculature during embryonic development, recapitulate a normal vascular phenotype as adults (Gu et al, 2005). Thus, the anti-angiogenic activity of Sema4A appears to be both temporally and spatially restricted.
In contrast to Sema4A, another class 4 semaphorin, Sema4D, induces endothelial cell proliferation and tubule formation via Plexin-B1 (Basile et al, 2004, 2005; Conrotto et al, 2005). The stimulatory effect of Sema4D may depend on the presence of c-Met, which was found to be a co-receptor for Plexin-B1 (Conrotto et al, 2005) and the associated RhoGEF (LARG)- (Basile et al, 2004) and phosphoinositide 3-kinase (PI3 kinase)-mediated signaling pathways (Basile et al, 2005). Our binding experiment showed that Sema4A also binds to Plexin-B1; however, the inhibitory effect of Sema4A was not suppressed when Plexin-B1 was knocked down using siRNA, indicating that Plexin-B1 is not the functional receptor for Sema4A. Sema4A may compete with Sema4D for Plexin-B1, but further study will be needed to resolve the way in which Sema4A and Sema4D exert opposing effects on endothelial cells.
VEGF binding to VEGFR2 leads to autophosphorylation of VEGFR2, after which the tyrosine-phosphorylated receptor recruits several adaptor proteins and exerts a variety of biological effects on endothelial cells (Zachary, 2003). In the present study, Sema4A failed to affect the binding of VEGF165 to VEGFR2 or the resulting tyrosine phosphorylation of the receptor, indicating that Sema4A does not compete with VEGF165 for VEGFR2. Instead, we found that Sema4A suppresses VEGF-induced phosphorylation of Akt and Rac-GTP production, both of which are downstream targets of PI3 kinase in the signal transduction pathway of VEGF/VEGFR2 (Missy et al, 1998; Eriksson et al, 2003; Zachary, 2003). Thus, Sema4A might suppress the PI3 kinase-dependent pathway.
Rac reportedly promotes VEGF-induced endothelial cell motility (Soga et al, 2001) and assembly of capillary tubes in vitro (Connolly et al, 2002), as well as integrin function at the leading edge of migrating endothelial cells (Kiosses et al, 2001). Integrin function is also involved in VEGF-mediated angiogenesis (Senger et al, 1997; Liu et al, 2003), and a number of endogenous inhibitors of angiogenesis, including endostatin, thrombospondin and tumstatin (Sund et al, 2005), as well as Sema3A (Serini et al, 2003), have been shown to mediate their anti-angiogenic effects via direct or indirect interactions with integrins. Taken together, these findings strongly support the notion that Sema4A suppresses VEGF-mediated vessel formation through sequential steps involving inhibition of the Rac pathway and β1-integrin function.
In summary, Sema4A is a potent anti-angiogenic molecule that suppresses VEGF-mediated migration and proliferation of endothelial cells by inhibiting Rac-mediated cytoskeletal rearrangement and integrin-mediated cell adhesion via Plexin-D1. Together with its activation of T-cell-mediated immune responses, our findings raise the possibility that Sema4A is a potentially useful therapeutic agent for the treatment of cancer.
Materials and methods
Genetically modified mice
The generation and genotyping of Sema4A-deficient mice was described previously (Kumanogoh et al, 2005).
Cell culture and reagents
HUVECs and endothelial basal medium containing endothelial cell growth supplements were purchased from Kurabo (Osaka, Japan). Recombinant human VEGF165 was from R&D Systems Inc. (Minneapolis, MN).
Construction of transfectants
Plexin family members were generated by RT–PCR and ligated into FLAG-CMV3 expression vectors (Sigma-Aldrich Corp., St Louis, MO). To generate mifepristone-dependent induction of Plexin-D1 expression in HEK293 cells, Plexin-D1 was ligated into pGene/V5-His expression vector (Invitrogen Corp., Carsbad, CA), and the resulting plasmids were transfected into HEK293 cells using Fugene-6 (Roche Molecular Biochemicals). Cell lines stably overexpressing the proteins encoded by these plasmids were established by selection with Zeocin (Invitrogen Corp., Carsbad, CA). An adenoviral vector system was used to analyze the gain-of-function of target proteins. For construction of adenoviral vectors containing constitutively active Rac (RacV12), dominant-negative Rac (RacN17), or GFP constructs were ligated into pShuttle2, after which the expression cassette was excised and ligated into BD Adeno-X viral DNA (BD Biosciences Clontech). After transfection of the adenoviral vector into HEK293 cells, adenovirus-containing medium was collected and concentrated. The incorporation of adenovirus into HUVECs was estimated to be over 90% using fluorescence microscopy.
Generation and production of soluble recombinant proteins
Sema4A-Fc, Sema4D-Fc and Sema3A-Fc were produced as described previously (Kumanogoh et al, 2000; Toyofuku et al, 2005). The nucleotide sequence encoding Met1 to E576 of Sema3E was amplified by PCR and inserted into pEF-Fc vector. The nucleotide sequence encoding the extracellular domain of Plexin-D1 (Plexin-D1-EC) (Met1 to Ala1260) was amplified by PCR and ligated into CMV-FLAG4 expression vector (Sigma-Aldrich Corp., St Louis, MO). After stably transfecting these vectors into P3U1 cells by electroporation, Sema3E-Fc and Flag-fused Plexin-D1-EC proteins were purified from serum-free medium conditioned by the transfectants using G-protein agarose and Flag-binding columns (Sigma-Aldrich Corp., St Louis, MO), respectively.
In vitro RNAi
siRNAs specific for control, Plexin-B1, B2, B3 and D1 were chemically synthesized and then transfected into HUVECs using OligofectAMINE Reagent (Invitrogen). The chosen sequences for human Plexin-B1, Plexin-B2, Plexin-B3 and Plexin-D1 siRNAs were: for Plexin-B1 siRNA, 5′-CUUCCUGUUCCAGAGCTT-3′ (sense) and 5′-GCUCAGCUGGAACAGGAAGTT-3′ (antisense); for Plexin-B2 siRNA, 5′-GCUGAUGCUGCGCAGGUCTT-3′ (sense) and 5′-AGACCUGUGCAGCAUCAGCTT-3′ (antisense); for Plexin-B3 siRNA, 5′-CCACUUGGCACUGGCACCUTT-3′ (sense) and 5′-AGGUGCCAGUGCCAAGUGGTT-3′ (antisense); for Plexin-D1 siRNA, 5′-GCACUUCCUCAUCGUCUUUTT-3′ (sense) and 5′-AAAGACGAUGAGGAAGUGCTT-3′ (antisense). After incubation for 16 h, the transfectants were subjected to migration assays.
HUVEC migration assays
HUVEC migration was assayed using modified Boyden chambers (Kurabo, Osaka, Japan). Growth factors in EB2 medium containing 1% FCS were placed in the bottom wells of the chambers. Polycarbonate filters with 8-μm pores were coated with 50 μg/ml fibronectin and 0.1% gelatin and placed between the test substances and the upper wells of the chambers. Cultured HUVECs were harvested by trypsinization, washed twice in DMEM, resuspended in DMEM containing 1% BSA, plated to a density of 2.5 × 105 cells/well in the upper wells of the chambers and incubated for 5 h at 37°C under a humidified atmosphere of 5% CO2/95% air. After the incubation period, a cotton ball was used to remove the cells that did not migrate from the upper side of the filters. The filters were then fixed with methanol, mounted onto microscope slides and stained with Giemsa solution. Cells that had migrated to the bottom side of the filters were visualized under a microscope (× 100 magnification) and counted. HUVECs infected with adenovirus containing Rac mutant constructs and GFP (multiplicity of infection 100) was used after 24 h post-infection.
HUVEC proliferation assays
HUVECs were plated to a density of 2 × 104 cells/cm2 on gelatinized 24-well plates and incubated for 12 h in EB2 medium (Kurabo, Osaka, Japan) supplemented with 2% FCS. Thereafter, the medium was changed, and growth factors and/or test substances were added. After 24 h, fresh medium and/or growth factors were added, and the cells were incubated for an additional 24 h. After washing the cells twice with PBS, their DNA was labeled with PicoGreen fluorescent reagent (Molecular Probes, Eugene, OR) and measured using a fluorescence spectrometer equipped with a microplate reader.
In vitro angiogenesis
Angiogenesis was assayed using a commercially available kit according to the manufacturer's instructions (Kurabo, Osaka, Japan). HUVECs were cultured for 11 days in the presence of VEGF165 (10 ng/ml) and test substances, after which they were fixed at room temperature in 70% ethanol. The fixed cells were then incubated first with mouse anti-human CD31 antibody (1:4000 dilution) for 1 h and then with a goat anti-mouse IgG AP-conjugated secondary antibody, which was visualization using 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium. The tube-like structures were measured in terms of total tube length/field (× 40) using NIH image software.
Cell adhesion assay
After serum starving HEK293 (1 × 104 cells) cells overnight, they were resuspended in 0.1 ml of DMEM with or without appropriate stimuli, plated on six-well plates coated with various extracellular matrices, incubated for 30 min at 37°C, washed three times with PBS, fixed in 8% glutaraldehyde and stained with 0.1% crystal violet in 20% methanol.
Chicken embryo cultures
Fertilized chicken eggs were incubated at 38°C in high humidity. Embryos, staged according to the Hamburger and Hamilton method (Hamburger and Hamilton, 1951), were removed from the eggs and placed ventral side up on collagen gel in culture dishes. To study the effect of Sema4A-Fc on angiogenesis gain of function, embryos were incubated with recombinant proteins and allowed to develop in a humidified incubator.
Chorioallantoic membrane assay
CAM assays were performed as described previously (Ribatti et al, 2000). Fertilized White Leghorn chick eggs (Takeuchi Farm Inc.) were incubated at 38°C under constant high humidity. On the third day of incubation, a square window was opened in the shell after removal of 2–3 ml of albumen, so as to detach the developing CAM from the shell. The window was then sealed with wrap of the same size, and the eggs were returned to the incubator. On day 8, 1 mm3 sterilized gelatin sponges (Gelfoam; Delasco, Council Bluffs, IA) that had been soaked in Sema4A-Fc dissolved in 3 μl of PBS, were implanted on the tops of the growing CAMs under sterile conditions. On day 10, the CAMs were examined and photographed in ovo under a Nikon stereomicroscope. Angiogenesis was estimated based on the angiogenic index (mean number of tertiary branch points seen under the experimental conditions minus the mean number of branch points seen in buffer controls with no angiogenic stimulator).
Corneal neovascularization assay
Corneal neovascularization was assayed as described previously (Stuart et al, 2003; Samolov et al, 2005). A single, penetrating, 8-0 silk suture was passed through the centers of the corneas of anesthetized mice. The subsequent neovascularation response was then studied until the entire cornea was invaded by pathologic vessels. The sutured eyes were enucleated after 3 or 8 days, immediately following injection of FITC-dextran (Sigma-Aldrich Inc., St Louis, MO, USA) into the left cardiac ventricle. After fixation, the corneas were excised, flat-mounted and photographed using a fluorescence microscope. The ratio of the area of the vascularized cornea to the total corneal area was calculated using the innermost vessel of the limbal arcade as the border.
Matrigel plug angiogenesis assays
Matrigel containing 50 ng/ml VEGF165 with or without Sema4A-Fc (10 μg/ml) was prepared and used as described previously for Matrigel plug angiogenesis assays (Angiolillo et al, 1995). Matrigel (0.6 ml/animal) was injected subcutaneously into female wild-type (BALB/c) or Sema4A knocked-out mice. After 7 days, the animals were killed, and the Matrigel plugs were removed, embedded in paraffin blocks and sectioned. The 5-μm-thick sections were then reacted with anti-CD31 antibody, an endothelial cell marker. CD31-positive blood vessels that had invaded the Matrigel were visualized, and microvessel density was measured as described previously (Sabo et al, 2001). All animal experiments were approved by the Ethics Committee of Osaka University.
Antibodies, immunoprecipitation and immunohistochemistry
We used rabbit polyclonal anti-factor VIII antibody (DAKO) and anti-Flk-1 antibody (VEGFR2) (Santa-Cruz Inc.), mouse monoclonal anti-phosphotyrosine antibody (4G10) (Santa-Cruz Inc.), anti-human β-integrin antibodies, HUTS-4 (β1-integrin stimulating antibody) and JBS5 (β1-integrin blocking antibody) (Chemicon). Immunoprecipitation was carried out according to the standard protocols. Whole-mount immunohistochemistry of chick embryo was carried out as described previously (Mason, 1998). Cytoskeletal rearrangement of HUVECs was estimated by phalloidin–rhodamine staining.
Surface biotinylation-chase experiment
HEK293 cells expressing Sema4A were biotinylated using a Cellular Labeling and Immunoprecipitation kit (Boehringer Mannheim). After biotinylation, the culture supernatants and the cells were harvested at the indicated times, washed and solubilized in 1% NP-40, after which they were centrifuged at 100 000 g for 1 h to remove membrane debris. The contents of the remaining supernatants and cell lysates were immunoprecipitated with anti-Sema4A antibody (Kumanogoh et al, 2005) and subjected to SDS–PAGE. The immunoblots were probed with streptavidin–POD conjugates and developed using ECL reagent (Amersham Pharmacia Biotech., Uppsala, Sweden).
In situ hybridization
Mouse embryos at the desired stages of development were fixed and then hybridized with DIG-labeled antisense RNA probes as described previously (Nieto et al, 1996). The cDNA sequences of Sema4A and Plexin-D1 were then used to transcribe cRNAs for in situ hybridization probes.
Rac activation assays
Because only activated, GTP-bound Rac binds to the CRIB domain of PAK (Sander et al, 1998), in vivo Rac1 activity was measured in pull-down assays using the GST-fused CRIB domain of PAK. Rac1 activation assay kits (Upstate Biotechnology Inc., Lake Placid, NY) were used in this study. To ensure consistent loading, total Rac1 protein also was measured by immunoblotting.
Radioiodination of VEGF165, Sema4A-Fc or Sema3E-Fc and chemical crosslinking experiments
The radioiodination of VEGF165 (50 ng/ml), Sema4A-Fc (50 nM) or Sema3E-Fc (5 nM) was achieved using IODO-BEADS (Pierce Biotechnology Inc., Rockford, IL), and specific activities ranging from 40 000 to 100 000 c.p.m./ng were obtained. HUVECs (3 × 106 cells) or HEK293 transfectants were incubated in binding buffer containing 125I-labeled VEGF165 or Sema4A-Fc for 2 h at 4°C. At the end of the experiment, ligand–receptor complexes on the cell surface were crosslinked with 0.5 mM disuccinimidyl suberate (Pierce), after which the crosslinked complexes were resolved by 6% SDS–PAGE, and the gels were dried and exposed to X-ray films. For the binding competition experiment, cells were incubated in binding buffer containing 125I-labeled Semaphorin-Fc and various amounts of unlabeled Semaphorin-Fc. After the crosslinking, extracts from cells were resolved by duplicate SDS–PAGE. One gel was subjected to the autoradiography, whereas another one was transferred to nitrocellulose and blotted membrane was immunoblotted with anti-Flag antibody to detect Plexin-D1.
Surface plasmon resonance assays using a BIAcore system
Binding experiments were carried out using surface plasmon resonance on a BIAcore instrument (BIAcore, Uppsala, Sweden). Anti-FLAG M2 antibody was covalently coupled via primary amine groups to the carboxylmethylated dextran matrix on a research grade CM5 sensor chip using an Amine Coupling kit. Recombinant Plexin-D1-FLAG protein was then injected and trapped on the anti-FLAG-coated sensor chip. Dissociation equilibrium constants (KD) were defined according to the equation KD=koff/kon and were estimated using BIAcore evaluation software.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
We thank Junko Yoshida and Tamiko Sugimoto for excellent technical assistance. We also thank Seiji Takashima for instruction of chemical crosslinking experiment and Hitoshi Kikutani for advice on preparation of manuscript. This study was supported by research grants from the Ministry of Education, Science, and Culture, Japan to TT and AK.
References
- Adams RH, Lohrum M, Klostermann A, Betz H, Puschel AW (1997) The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. EMBO J 16: 6077–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angiolillo AL, Sgadari C, Taub DD, Liao F, Farber JM, Maheshwari S, Kleinman HK, Reaman GH, Tosato G (1995) Human interferon-inducible protein 10 is a potent inhibitor of angiogenesis in vivo. J Exp Med 182: 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham HK, Lee TH, Koh Y, Kim TA, Jiang S, Sussman M, Samarel AM, Avraham S (2003) Vascular endothelial growth factor regulates focal adhesion assembly in human brain microvascular endothelial cells through activation of the focal adhesion kinase and related adhesion focal tyrosine kinase. J Biol Chem 278: 36661–36668 [DOI] [PubMed] [Google Scholar]
- Basile JR, Afkhami T, Gutkind JS, Barac A, Zhu T, Guan KL (2005) Semaphorin 4D/plexin-B1 induces endothelial cell migration through the activation of PYK2, Src, and the phosphatidylinositol 3-kinase–Akt pathway. Class IV semaphorins promote angiogenesis by stimulating Rho-initiated pathways through plexin-B. Mol Cell Biol 25: 6889–6898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile JR, Barac A, Zhu T, Guan KL, Gutkind JS (2004) Class IV semaphorins promote angiogenesis by stimulating Rho-initiated pathways through plexin-B. Cancer Res 64: 5212–5224 [DOI] [PubMed] [Google Scholar]
- Bielenberg DR, Hida Y, Shimizu A, Kaipainen A, Kreuter M, Kim CC, Klagsbrun M (2004) Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J Clin Invest 114: 1260–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K (2004) Rho and Rac take center stage. Cell 116: 167–179 [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Tessier-Lavigne M (2005) Common mechanisms of nerve and blood vessel wiring. Nature 436: 193–200 [DOI] [PubMed] [Google Scholar]
- Chen H, He Z, Bagri A, Tessier-Lavigne M (1998) Semaphorin–neuropilin interactions underlying sympathetic axon responses to class III semaphorins. Neuron 21: 1283–1290 [DOI] [PubMed] [Google Scholar]
- Christensen C, Ambartsumian N, Gilestro G, Thomsen B, Comoglio P, Tamagnone L, Guldberg P, Lukanidin E (2005) Proteolytic processing converts the repelling signal Sema3E into an inducer of invasive growth and lung metastasis. Cancer Res 65: 6167–6177 [DOI] [PubMed] [Google Scholar]
- Connolly JO, Simpson N, Hewlett L, Hall A (2002) Rac regulates endothelial morphogenesis and capillary assembly. Mol Biol Cell 13: 2474–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrotto P, Valdembri D, Corso S, Serini G, Tamagnone L, Comoglio PM, Bussolino F, Giordano S (2005) Sema4D induces angiogenesis through Met recruitment by Plexin B1. Blood 105: 4321–4329 [DOI] [PubMed] [Google Scholar]
- Eichmann A, Le Noble F, Autiero M, Carmeliet P (2005) Guidance of vascular and neural network formation. Curr Opin Neurobiol 15: 108–115 [DOI] [PubMed] [Google Scholar]
- Eriksson A, Cao R, Roy J, Tritsaris K, Wahlestedt C, Dissing S, Thyberg J, Cao Y (2003) Small GTP-binding protein Rac is an essential mediator of vascular endothelial growth factor-induced endothelial fenestrations and vascular permeability. Circulation 107: 1532–1538 [DOI] [PubMed] [Google Scholar]
- Gitler AD, Lu MM, Epstein JA (2004) PlexinD1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Dev Cell 7: 107–116 [DOI] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, Ginty DD (2003) Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell 5: 45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Yoshida Y, Livet J, Reimert DV, Mann F, Merte J, Henderson CE, Jessell TM, Kolodkin AL, Ginty DD (2005) Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science 307: 265–268 [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL (1951) A series of normal stages in the development of the chick embryo. J Morph 88: 49–92 [PubMed] [Google Scholar]
- Hynes RO (2002) A reevaluation of integrins as regulators of angiogenesis. Nat Med 8: 918–921 [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, Fujisawa H (1999) A requirement for neuropilin-1 in embryonic vessel formation. Development 126: 4895–4902 [DOI] [PubMed] [Google Scholar]
- Kessler O, Shraga-Heled N, Lange T, Gutmann-Raviv N, Sabo E, Baruch L, Machluf M, Neufeld G (2004) Semaphorin-3F is an inhibitor of tumor angiogenesis. Cancer Res 64: 1008–1015 [DOI] [PubMed] [Google Scholar]
- Kiosses WB, Shattil SJ, Pampori N, Schwartz MA (2001) Rac recruits high-affinity integrin alphavbeta3 to lamellipodia in endothelial cell migration. Nat Cell Biol 3: 316–320 [DOI] [PubMed] [Google Scholar]
- Klostermann A, Lohrum M, Adams RH, Puschel AW, Betz H (1998) The chemorepulsive activity of the axonal guidance signal semaphorin D requires dimerization. The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. J Biol Chem 273: 7326–7331 [DOI] [PubMed] [Google Scholar]
- Kolodkin AL, Levengood DV, Rowe EG, Tai YT, Giger RJ, Ginty DD (1997) Neuropilin is a semaphorin III receptor. Cell 90: 753–762 [DOI] [PubMed] [Google Scholar]
- Kolodkin AL, Matthes DJ, Goodman CS (1993) The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell 75: 1389–1399 [DOI] [PubMed] [Google Scholar]
- Koppel AM, Raper JA (1998) Collapsin-1 covalently dimerizes, and dimerization is necessary for collapsing activity. J Biol Chem 273: 15708–15713 [DOI] [PubMed] [Google Scholar]
- Korff T, Augustin HG (1998) Integration of endothelial cells in multicellular spheroids prevents apoptosis and induces differentiation. J Cell Biol 143: 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumanogoh A, Marukawa S, Suzuki K, Takegahara N, Watanabe C, Ch'ng E, Ishida I, Fujimura H, Sakoda S, Yoshida K, Kikutani H (2002) Class IV semaphorin Sema4A enhances T-cell activation and interacts with Tim-2. Nature 419: 629–633 [DOI] [PubMed] [Google Scholar]
- Kumanogoh A, Shikina T, Suzuki K, Uematsu S, Yukawa K, Kashiwamura S, Tsutsui H, Yamamoto M, Takamatsu H, Ko-Mitamura EP, Takegahara N, Marukawa S, Ishida I, Morishita H, Prasad DV, Tamura M, Mizui M, Toyofuku T, Akira S, Takeda K, Okabe M, Kikutani H (2005) Nonredundant roles of Sema4A in the immune system: defective T cell priming and Th1/Th2 regulation in Sema4A-deficient mice. Immunity 22: 305–316 [DOI] [PubMed] [Google Scholar]
- Kumanogoh A, Watanabe C, Lee I, Wang X, Shi W, Araki H, Hirata H, Iwahori K, Uchida J, Yasui T, Matsumoto M, Yoshida K, Yakura H, Pan C, Parnes JR, Kikutani H (2000) Identification of CD72 as a lymphocyte receptor for the class IV semaphorin CD100: a novel mechanism for regulating B cell signaling. Immunity 13: 621–631 [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Snyder R, Soma A, Shirakawa T, Ziober BL, Fairman RM, Herlyn M, Velazquez OC (2003) VEGF-A and alphaVbeta3 integrin synergistically rescue angiogenesis via N-Ras and PI3-K signaling in human microvascular endothelial cells. FASEB J 17: 1931–1933 [DOI] [PubMed] [Google Scholar]
- Luo Y, Raible D, Raper JA (1993) Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell 75: 217–227 [DOI] [PubMed] [Google Scholar]
- Mason I (1998) Immunohistochemistry on whole embryo. In Molecular Embryology: Methods and Protocols, Sharpe PT, Mason I (eds), pp 663–666. NJ, USA: Humana Press, Inc [Google Scholar]
- Miao HQ, Soker S, Feiner L, Alonso JL, Raper JA, Klagsbrun M (1999) Neuropilin-1 mediates collapsin-1/semaphorin III inhibition of endothelial cell motility: functional competition of collapsin-1 and vascular endothelial growth factor-165. J Cell Biol 146: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missy K, Van Poucke V, Raynal P, Viala C, Mauco G, Plantavid M, Chap H, Payrastre B (1998) Lipid products of phosphoinositide 3-kinase interact with Rac1 GTPase and stimulate GDP dissociation. J Biol Chem 273: 30279–30286 [DOI] [PubMed] [Google Scholar]
- Nieto MA, Patel K, Wilkinson DG (1996) In situ hybridization analysis of chick embryos in whole mount and tissue sections. Methods Cell Biol 51: 219–235 [DOI] [PubMed] [Google Scholar]
- Nikolopoulos SN, Blaikie P, Yoshioka T, Guo W, Giancotti FG (2004) Integrin beta4 signaling promotes tumor angiogenesis. Cancer Cell 6: 471–483 [DOI] [PubMed] [Google Scholar]
- Ribatti D, Vacca A, Roncali L, Dammacco F (2000) The chick embryo chorioallantoic membrane as a model for in vivo research on anti-angiogenesis. Curr Pharm Biotechnol 1: 73–82 [DOI] [PubMed] [Google Scholar]
- Sabo E, Boltenko A, Sova Y, Stein A, Kleinhaus S, Resnick MB (2001) Microscopic analysis and significance of vascular architectural complexity in renal cell carcinoma. Clin Cancer Res 7: 533–537 [PubMed] [Google Scholar]
- Samolov B, Steen B, Seregard S, van der Ploeg I, Montan P, Kvanta A (2005) Delayed inflammation-associated corneal neovascularization in MMP-2-deficient mice. Exp Eye Res 80: 159–166 [DOI] [PubMed] [Google Scholar]
- Sander EE, van Delft S, ten Klooster JP, Reid T, van der Kammen RA, Michiels F, Collard JG (1998) Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell–cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol 143: 1385–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger DR, Claffey KP, Benes JE, Perruzzi CA, Sergiou AP, Detmar M (1997) Angiogenesis promoted by vascular endothelial growth factor: regulation through alpha1beta1 and alpha2beta1 integrins. Proc Natl Acad Sci USA 94: 13612–13617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serini G, Valdembri D, Bussolino F (2006) Integrins and angiogenesis: a sticky business. Exp Cell Res 312: 651–658 [DOI] [PubMed] [Google Scholar]
- Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, Zammataro L, Primo L, Tamagnone L, Logan M, Tessier-Lavigne M, Taniguchi M, Puschel AW, Bussolino F (2003) Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature 424: 391–397 [DOI] [PubMed] [Google Scholar]
- Soga N, Namba N, McAllister S, Cornelius L, Teitelbaum SL, Dowdy SF, Kawamura J, Hruska KA (2001) Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp Cell Res 269: 73–87 [DOI] [PubMed] [Google Scholar]
- Soker S, Miao HQ, Nomi M, Takashima S, Klagsbrun M, Feiner L, Alonso JL, Raper JA, Neufeld G (2002) VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J Cell Biochem 85: 357–368 [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M (1998) Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92: 735–745 [DOI] [PubMed] [Google Scholar]
- Stuart PM, Pan F, Plambeck S, Ferguson TA (2003) FasL-Fas interactions regulate neovascularization in the cornea. Invest Ophthalmol Vis Sci 44: 93–98 [DOI] [PubMed] [Google Scholar]
- Sund M, Hamano Y, Sugimoto H, Sudhakar A, Soubasakos M, Yerramalla U, Benjamin LE, Lawler J, Kieran M, Shah A, Kalluri R (2005) Function of endogenous inhibitors of angiogenesis as endothelium-specific tumor suppressors. Proc Natl Acad Sci USA 102: 2934–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Vazquez J, Gitler AD, Fraser SD, Berk JD, Van NP, Fishman MC, Childs S, Epstein JA, Weinstein BM (2004) Semaphorin–plexin signaling guides patterning of the developing vasculature. Dev Cell 7: 117–123 [DOI] [PubMed] [Google Scholar]
- Toyofuku T, Yoshida J, Sugimoto T, Zhang H, Kumanogoh A, Hori M, Kikutani H (2005) FARP2 triggers signals for Sema3A-mediated axonal repulsion. Nat Neurosci 8: 1712–1719 [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J (2000) Vascular-specific growth factors and blood vessel formation. Nature 407: 242–248 [DOI] [PubMed] [Google Scholar]
- Zachary I (2003) VEGF signalling: integration and multi-tasking in endothelial cell biology. Biochem Soc Trans 31: 1171–1177 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5