

Abstract
The present study quantified the cleaved form of the microtubule associated protein tau (cleaved MAP-tau, C-tau), a previously demonstrated marker of CNS toxicity, following the administration of monoamine-depleting regimens of the psychostimulant drugs amphetamine (AMPH), methamphetamine (METH), ±3,4-methylenedioxymethamphetamine (MDMA), or para-methoxyamphetamine (PMA) in an attempt to further characterize psychostimulant-induced toxicity. A dopamine (DA)-depleting regimen of AMPH produced an increase in C-tau immunoreactivity in the striatum, while a DA- and serotonin (5-HT)-depleting regimen of METH produced an increase in the number of C-tau immunoreactive cells in the striatum and CA2/CA3 and dentate gyrus regions of the hippocampus. MDMA and PMA, two psychostimulant drugs that produce selective 5-HT depletion in the striatum, had no effect on C-tau immunoreactivity in the striatum or hippocampus. Furthermore, 5,7-dihydroxytryptamine (5,7-DHT), an established 5-HT selective neurotoxin, did not produce an increase in C-tau immunoreactivity. Dual fluorescent immunocytochemistry with antibodies to GFAP and C-tau indicated that C-tau immunoreactivity was present in astrocytes, not neurons, suggesting that increased C-tau may be an alternative indicator of reactive gliosis. The present results are consistent with previous findings that the DA-depleting psychostimulants AMPH and METH produce reactive gliosis whereas the 5-HT-depleting drugs MDMA and PMA, as well as the known 5-HT selective neurotoxin 5,7-DHT, do not produce an appreciable glial response.
Keywords: methamphetamine; 3,4-methylenedioxymethamphetamine; amphetamine; para-methoxyamphetamine; 5,7-dihydroxytryptamine; toxicity
Methamphetamine (METH) and ±3,4-methylenedioxymethamphetamine (MDMA) are substituted phenylethylamines whose popularity among young adults continues to be a public health concern. Results from studies in humans, non-human primates and rodents support the view that these compounds cause structural damage to monoamine nerve terminals and are thereby neurotoxic (O’Callaghan, 2002a, Lyles and Cadet, 2003, Itzhak and Achat-Mendes, 2004). Such data include the finding that repeated high-dose administration of METH results in a reduction in striatal dopamine (DA) and serotonin (5-HT) concentrations (Kogan et al., 1976, Ricaurte et al., 1980, Wagner et al., 1980), a decrease in the activity of tyrosine and tryptophan hydroxylase, the rate-limiting enzymes responsible for the production of DA and 5-HT (Hotchkiss and Gibb, 1980), and reduced number of amine uptake sites (Wagner et al., 1980). Likewise, repeated high-dose administration of MDMA produces selective, long-term reductions in striatal 5-HT concentration (Schmidt, 1987, Battaglia et al., 1988), tryptophan hydroxylase activity (Stone et al., 1986, Schmidt and Taylor, 1987, Stone et al., 1987) and the number of 5-HT reuptake sites (Battaglia et al., 1987).
Additional biomarkers support the assertion that METH produces neurotoxicity; these include increased glial fibrillary acidic protein (GFAP) expression and immunoreactivity (O’Callaghan and Miller, 1993, Bowyer et al., 1994) and increased silver staining for degenerating neurons (Ricaurte et al., 1982, Bowyer et al., 1994). Newer markers, including Fluoro-Jade staining (Schmued and Bowyer, 1997, Eisch and Marshall, 1998, Eisch et al., 1998), as well as microglial activation (Thomas et al., 2004, Thomas and Kuhn, 2005), increased density of peripheral benzodiazepine receptors and increased expression of heat shock protein 27 (both markers of gliosis) (Pubill et al., 2003), are also in support of METH-induced neurotoxicity.
However, MDMA appears to be less effective when compared to METH in producing non-serotonergic markers of neurotoxicity. For example, MDMA appears to be relatively ineffective in producing reactive gliosis, as indicated by a lack of a pronounced increase in expression or immunoreactivity of GFAP (O’Callaghan and Miller, 1993, Pubill et al., 2003, Wang et al., 2004b), although in a single exception, Aguirre and colleagues report an increase in GFAP immunoreactivity in the hippocampus 7 days after MDMA administration (Aguirre et al., 1999). MDMA also does not produce an appreciable increase in the expression of heat shock protein 27, particularly when compared to responses evoked by METH (Pubill et al., 2003). MDMA does not consistently produce increases in markers of gliosis, as Orio et al. (2004) demonstrate early increases in IL-1β and ipheral benzodiazepine receptor (PBZ) binding in theper hypothalamus and cortex, yet Pubill et al. (2003) indicate that MDMA does not produce increased microglial activation as indicated by [3H] PK11195 binding or OX-6 immunostaining in the striatum and cortex. Wang et al (2004b) have reported that SERT expression remains unchanged two weeks after a multiple-dose regimen of MDMA despite studies that have decreased radioligand binding to SERT. However, these results are contradicted by a recent report of Xie et al. (Xie et al., 2006) in which a MDMA-induced reduction in SERT protein expression was demonstrated, suggesting a loss of 5-HT axons. Additionally, studies have indicated a lack of Fluoro-Jade-B positive neurons in the hippocampus and striatum following administration of MDMA, although a limited number of degenerating neurons have been noted in other brain regions (Schmued, 2003). Furthermore, although some investigators have reported silver staining of degenerating axon terminals in the brains of rats treated with MDMA (Commins et al., 1987, Molliver et al., 1990), others have reported inconsistent results or patterns of staining that are not in agreement with other indicators of altered 5-HT innervation (Jensen et al., 1993). Given the mixed data regarding loss of 5-HT nerve terminals following administration of MDMA, investigation of additional markers of CNS injury are warranted.
Recently, proteolytically cleaved MAP-tau has been reported to be a novel biomarker of CNS toxicity (Zemlan et al., 1999, Irazuzta et al., 2001). MAP-tau is primarily a cytoskeletal protein which functions to stabilize microtubules. Upon CNS damage by a variety of insults, MAP-tau appears to be proteolytically cleaved (Irazuzta et al., 2001, Zemlan et al., 2002). Cleaved tau (C-tau) concentrations have been shown to be increased following administration of the prototypic neurotoxins kainic acid (Zemlan et al., 2003) and trimethyltin (Straiko et al., 2005 (submitted)) or following traumatic brain injury in rodents (Gabbita et al., 2005) and humans (Zemlan et al., 2002). Notably, METH also has been reported to produce a time-dependent increase in C-tau in the striatum, hippocampus and, to a lesser extent, frontal cortex (Wallace et al., 2003). The purpose of the present study was to compare the extent of C-tau formation produced by monoamine-depleting regimens of METH, MDMA, amphetamine (AMPH) and para-methoxyamphetamine (PMA).
Experimental Procedures
Animals and Treatment Procedures
Adult male (270–300 g) Sprague-Dawley rats (Harlan Laboratories; Indianapolis, IN, USA) were group-housed, maintained on a 12 hour light/dark cycle and allowed food and water ad libitum. Dosing regimens for drugs were as follows: AMPH (iprindole, 10 mg/kg, i.p. followed 60 minutes later with AMPH, 9 mg/kg, i.p.); METH (a total of 4 injections consisting of 2 i.p. injections of 10 mg/kg, and 1 i.p. injection each of 7.5 mg/kg and 5 mg/kg spaced 2 hours apart); MDMA (10 mg/kg, i.p., given every 2 hours for 4 injections) or PMA (10 mg/kg, i.p., given every 2 hours for 4 injections). All drugs were injected in a volume of 1 ml/kg. Vehicle injections consisted of 0.9% NaCl. Two hours prior to and throughout drug treatment, rats were maintained at an elevated room temperature of 24–25 ºC.
In separate experiments, rats received i.c.v. injections (coordinates: +0.8 mm A/P; 1.4 mm L from Bregma, 3.5 mm ventral to the surface of the brain) of 10 μl of vehicle (0.1% ascorbic acid in 0.9% NaCl) or 5,7-dihydroxytryptamine (5,7-DHT) (150 μg) 60 minutes following the administration of mazindol (5 mg/kg, i.p.).
Effort was made to minimize the number of animals used in this study. Animal procedures adhered to the policies set forth in the NRC Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee.
Dopamine and Serotonin Analyses
Three days following the administration of drug or vehicle, rats were killed by decapitation and brains removed. Striatum was dissected from 1 mm coronal sections of the brain and maintained at −80 ºC until analysis for dopamine (DA) and serotonin (5-HT). For animals receiving i.c.v. injections, the striatum ipsilateral to the injection site was analyzed. DA and 5-HT concentrations were determined by high performance liquid chromatography with electrochemical detection (Gudelsky et al., 1994). The mobile phase consisted of 35 mM citric acid, 54 mM sodium acetate, 60 mg/L octanesulfonic acid, 50 mg/L EDTA, 5% (volume/volume) methanol at pH 4.2. The mobile phase was pumped at 0.3 mL/min onto a C18 column connected to an LC-4C amperometric detector (BAS, Inc., West Lafayette, IN, USA). Peak heights were quantified with a Hewlett-Packard integrator.
Tissue Preparation for C-tau Analysis
Rats were treated with drug or vehicle and, 3 days later, deeply anesthetized with sodium pentobarbital (200 mg/kg, i.p.) and perfused transcardially with 200 ml 0.9% NaCl followed by 500 ml 4% paraformaldehyde made in PB (0.1 M, pH 7.3). Following perfusion, brains were removed and postfixed in 4% paraformaldehyde for 1 hour, then placed in 20% sucrose and stored at 4 ºC until the time of sectioning. Coronal sections were cut at 35 μm thickness in 4 parallel series on a freezing microtome from the olfactory region through the rostral brainstem and stored as floating sections in cryoprotectant (30% sucrose, 30% ethylene glycol in 0.1 M PB; (Watson et al., 1986)) at −20 ºC until the time of staining.
C-tau Immunocytochemistry
All staining was performed at room temperature in the light. Free-floating sections were rinsed in 0.1 M PBS for 4 hours, changing buffer every 10 minutes, to remove cryoprotectant and then in TBST (TBS containing 1% Tween, 3 rinses of 10 minutes each) and then blocked in 0.5% H2O2 in PBS for 20 minutes. Sections were rinsed in TBST and blocked a second time in 4% normal horse serum for 1 hour and incubated overnight in PBS containing the primary antibody C-tau 7 (1:120,000). Following washes in TBST, sections were incubated in secondary antibody (biotinlyated anti-mouse IgG; Vector Laboratories, Burlingame, CA, USA) for 1 hour, followed by rinsing in TBST. Sections were incubated in Vectastain ABC-HRP reagent (Vector Laboratories) for 30 minutes and bound antibody visualized with nickel sulfate enhanced diaminobenzidine (DAB, Sigma, St. Louis, MO, USA). Sections were mounted on glass slides with 0.3% gelatin, air-dried overnight, rehydrated in ddH20 and then dehydrated in graded alcohol and coverslipped with DPX (Electron Microscopy Sciences, Fort Washington, PA, USA) (Balfour et al., 2006).
C-tau and GFAP Dual Fluorescent Immunocytochemistry
All staining was performed at room temperature in the light unless indicated otherwise. Free-floating sections were thoroughly washed in 10 minute rinses of 0.1 M PBS containing 1% Tween for 4 hours to remove cryoprotectant, then washed in TBST and blocked in 4% normal horse serum in TBS for 1 hour. Sections were incubated overnight in TBS containing the primary antibodies C-tau7 (1:1000) and rabbit anti-GFAP (1:250; Sigma). Following washes in TBST, tissue sections were covered with aluminum foil to prevent exposure to the light. Sections were incubated in CY3-conjugated donkey anti-mouse (1:100; Jackson ImmunoResearch, Westgrove, PA, USA) for 30 minutes, rinsed with TBS and incubated in Alexa 488-conjugated goat anti-rabbit (1:100; Molecular Probes, Eugene, OR, USA) for 30 minutes. Sections were rinsed in phosphate buffer, mounted on glass slides and coverslipped with Gelvatol (Harlow and Lane, 1988) containing an anti-fading agent (1,4-diazabicyclo(2,2)octane; Sigma).
Data Analysis
Camera lucida drawings were made of each analyzed section from each rat using a Leica microscope with an attached drawing tube (Leica Microsystems; Wetzlar, Germany). In the striatum of each vehicle- or drug-treated rat, cell counts were made in standardized areas (0.36 mm2) in 4 bilateral sections approximately 280 μm apart representative of 4 rostral to caudal levels ranging from Bregma +1.20 to Bregma +0.48. In the hippocampus of METH-, MDMA- or VEH-treated rats, cell counts were made in standardized areas of the CA1 (0.4 mm2), CA2/CA3 (0.36 mm2) and dentate gyrus (0.4 mm2), in four bilateral sections approximately 280 μm apart, representative of 4 rostral to caudal levels of the dorsal hippocampus ranging from Bregma −2.56 to Bregma −3.30. The average cell count of the four striatal and hippocampal sections were determined for each animal. In rats treated with VEH or 5,7-DHT, cell counts were determined in standard areas (0.36 mm2) in four sections of striatum ipsilateral to the injection site approximately 280 μm apart. For each area of analysis, the average of the four sections was determined per animal and group averages were calculated.
Digital images of immunostained sections were captured using a digital camera (Magnafire, Optronics) attached to a Leica microscope (Leica Microsystems; Wetzlar, Germany). To demonstrate the location of C-tau and GFAP, fluorescent-stained sections were examined using a Zeiss laser-scanning confocal microscope system (Zeiss LSM-510; Heidelburg, Germany). CY3 fluorescence was imaged with a 567 nm emission filter and a HeNe laser, and Alexa 488 with a 505 nm emission filter and Argon laser. Stacks of 5 μm optical sections along the z-axis were obtained at 25x magnification. Images were imported into Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) to compose figures and were only adjusted for brightness.
Statistical Analyses
Analysis of the number of C-tau positive cells and tissue concentrations of DA and 5-HT were evaluated with a one-way analysis of variance. Individual post-hoc group comparisons were made using the Student-Newman-Keuls test. Treatment differences were considered significant at P< 0.05.
Results
Concentrations of DA and/or 5-HT in the striatum of rats 3 days following the regimens of METH, AMPH, MDMA, and PMA are documented in Table 1. Treatment with either AMPH or METH produced a significant [F(4,44) = 45.039; P < 0.001] decrease in striatal DA concentrations, whereas the striatal DA concentration was not significantly affected by treatment with MDMA (P = 0.190) or PMA (P = 0.240) compared to VEH-treated controls. In contrast, treatment with METH, MDMA or PMA produced a significant [F(4,44) = 6.982; P < 0.001] decrease in striatal concentrations of 5-HT compared to VEH-treated controls. The striatal concentration of 5-HT was not significantly affected by AMPH (P = 0.203). Treatment with 5,7-DHT produced a significant [F(1,8) = 12.822; P < 0.01] decrease in striatal 5-HT concentrations compared to VEH-injected controls but was without effect on striatal DA concentrations (P = 0.144) (Table 3).
Table 1.
Striatal dopamine and serotonin depletions produced by the psychostimulant drugs amphetamine, methamphetamine, ± 3,4 methylenedioxymethamphetamine, or para-methoxyamphetamine. Concentrations of dopamine (DA) and serotonin (5-HT) in the striatum of rats were determined 3 days after treatment with vehicle (saline, i.p.), amphetamine (AMPH; iprindole (10 mg/kg, i.p.) followed 1 hour later by AMPH (9.2 mg/kg, i.p.)) methamphetamine (METH; a total of 4 injections consisting of 2 i.p. injections of 10 mg/kg, and 1 i.p. injection each of 7.5 mg/kg and 5 mg/kg spaced 2 hours apart), ±3,4-methylenedioxymethamphetamine (MDMA; 10 mg/kg, i.p., given every 2 hours for 4 injections), or para-methoxyamphetamine (PMA; 10 mg/kg, i.p. given every two hours for four injections). Values represent the mean ± SEM. Numbers in parentheses equals the number of determinations.
| Drug | DA (ng/mg tissue ± SEM) | 5-HT (ng/mg tissue ± SEM) |
|---|---|---|
| Vehicle | 11.9 ± 0.43 (17) | 0.41 ± 0.03 (17) |
| AMPH | 2.2 ± 0.80*** (6) | 0.36 ± 0.04 (6) |
| METH | 4.2 ± 0.79*** (7) | 0.26 ± 0.03*** (7) |
| MDMA | 10.3 ± 0.83 (7) | 0.26 ± 0.02** (7) |
| PMA | 10.8 ± 0.62 (7) | 0.27 ± 0.03** (7) |
P < 0.001 and
P < 0.01 compared to vehicle-treated rats.
Table 3.
Dopamine and 5-HT concentrations and C-tau immunoreactive cells in the striatum of rats treated with 5,7-DHT. Concentrations of DA and 5-HT were determined and number of C-tau positive cells were counted in defined regions of the striatum of rats killed 3 days after i.c.v. administration of vehicle or 5,7-DHT (150 μg/10 μl). Values represent mean ± SEM of 3–6 rats.
| Drug | DA | 5-HT | Striatal C-tau |
|---|---|---|---|
| (ng/mg tissue ± SEM) | (cell count ± SEM) | ||
| Vehicle | 16.5 ± 0.59 | 0.52 ± 0.12 | 2.58 ± 0.30 |
| 5,7-DHT | 14.6 ± 0.84 | 0.15 ± 0.04** | 3.56 ± 2.23 |
P < 0.01 compared to vehicle-treated controls.
The numbers of C-tau immunoreactive cells in the striatum and CA1, CA2/CA3 and dentate gyrus regions of the hippocampus of rats treated with VEH, METH or MDMA are depicted in Figure 1. Treatment with METH resulted in a number of C-tau immunoreactive cells in the striatum that was 19 times greater than that in VEH-treated controls [F(2,17) = 64.607; P<0.001]. METH also produced a significant increase in the number of C-tau immunoreactive cells in the CA2/CA3 [F(2,17) = 7.180; P < 0.05] and dentate gyrus [F(2,17) = 5.646; P < 0.05] regions of the hippocampus. The number of C-tau positive cells in the CA2/CA3 and dentate gyrus regions of the hippocampus of METH-treated rats was almost twice that in the same brain region of VEH-treated rats. The number of C-tau immunoreactive cells in the CA1 region of the hippocampus was not significantly different between VEH- and METH-treated rats, although there was a trend for a METH-induced increase [F(2,17) = 6.155; P = 0.069]. In contrast to the significant METH-induced increase in C-tau formation, MDMA treatment did not significantly alter the number of C-tau positive cells in the striatum (P = 0.922), CA1 (P = 0.247), CA2/CA3 (P = 0.329) or dentate gyrus (P = 0.696) regions of the hippocampus (Figure 1). Photomicrographs of sections of striatum (Figure 2A, C, E) and dentate gyrus (Figure 2B, D, F) of rats treated with VEH (Figure 2A, B) or monoamine-depleting regimens of METH (Figure 2C, D) or MDMA (Figure 2E, F) further illustrate the increase in C-tau immunoreactive cells present in the brain regions in METH-, but not MDMA-, treated rats.
Figure 1.
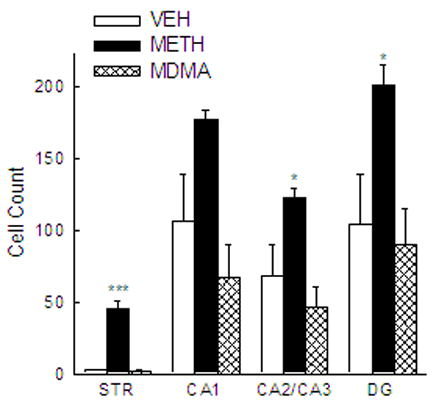
Quantification of C-tau labeling in the striatum and hippocampus of rats treated with methamphetamine or ± 3,4 methylenedioxymethamphetamine. C-tau positive cells were counted in defined regions of the striatum and CA1, CA2/CA3 and dentate gyrus regions of the hippocampus of rats killed 3 days after treatment with METH, MDMA or VEH, administered as described in the legend of Table 1. Bars represent mean ± SEM of 5–9 rats. *** P < 0.001 and * P < 0.05 compared to VEH-treated rats.
Figure 2.
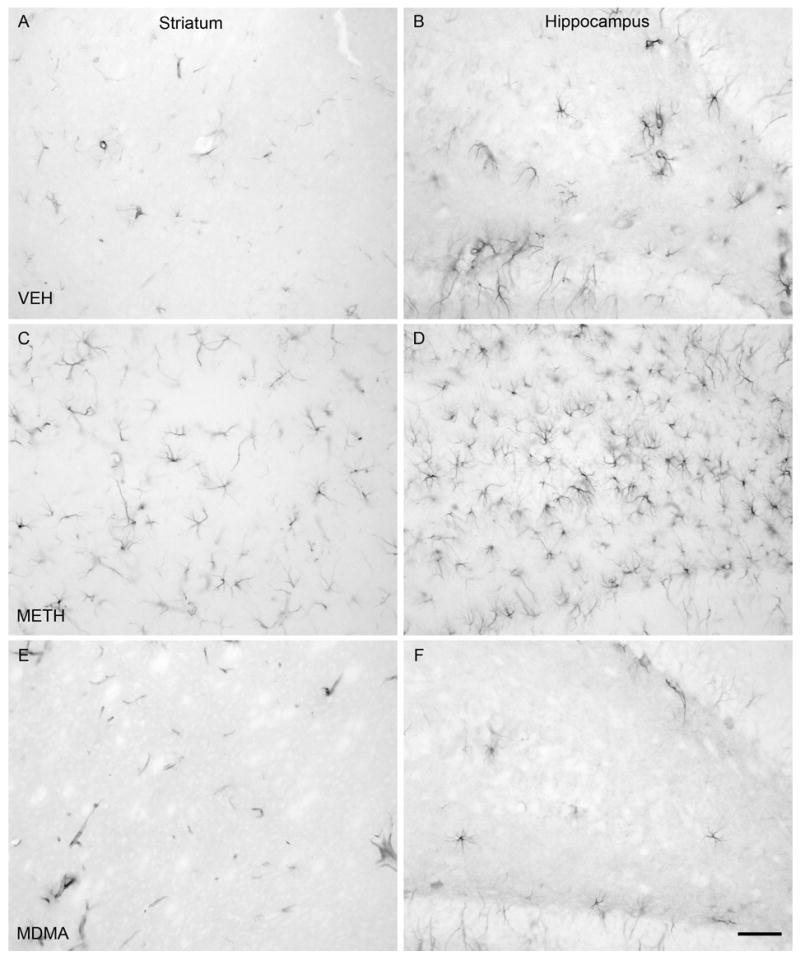
C-tau immunolabeling in the striatum and dentate gyrus regions of the hippocampus. Immunocytochemistry was performed with an antibody specific to cleaved MAP-tau (C-tau7) and striatal (panels A, C, E) and hippocampal (panels B, D, F) tissue of rats killed 3 days after treatment with VEH (panels A and B), METH (panels C and D) or MDMA (panels E and F). Drugs were administered as described in the legend of Table 1. Magnification, 20x.
C-tau immunoreactive cells also were quantified in the striatum of rats treated with VEH or dosing regimens of AMPH or PMA demonstrated to produce depletion of DA alone (AMPH) or 5-HT alone (PMA) (Table 1). Although AMPH treatment resulted in a significant increase in C-tau immunoreactive cells [F(2,11) = 6.859; P < 0.05] in the striatum compared to VEH-treated controls, PMA treatment was without effect in this regard (P = 0.461) (Table 2).
Table 2.
Quantification of C-tau labeling in the striatum of rats treated with amphetamine or para-methoxyamphetamine. C-tau positive cells were counted in defined regions of the striatum of rats killed 3 days after injection with VEH, AMPH or PMA as described in the legend of Table 1. Values represent mean ± SEM of 4–5 rats.
| Drug | Striatal C-tau (cell count ± SEM) |
|---|---|
| Vehicle | 17 ± 6 |
| AMPH | 51 ± 7* |
| PMA | 24 ± 8 |
P < 0.05 compared to vehicle-treated rats.
Additionally, C-tau immunoreactive cells were quantified in the striatum of rats that received i.c.v. injections of VEH or 5,7-DHT, a selective 5-HT neurotoxin. Although treatment with 5,7-DHT produced a 70% reduction in the striatal concentration of 5-HT, 5,7-DHT did not significantly increase the number of C-tau immunoreactive cells in the striatum (P = 0.727) (Table 3).
Additional studies employing dual fluorescent immunocytochemistry with antibodies for C-tau and GFAP (an astrocytic marker) and confocal microscopy were performed to examine the potential localization of C-tau within astrocytes. Cells in the striatum (Figure 3A, C and E) and dentate gyrus (Figure 3B, D and F) of VEH- (Figure 3A, B), METH- (Figure 3C, D) and MDMA- (Figure 3E, F) treated rats that were C-tau immunoreactive were also immunoreactive for GFAP. As indicated in Figure 3, there was essentially complete overlap of C-tau and GFAP immunoreactivity, indicating that C-tau positive cells were astrocytes.
Figure 3.
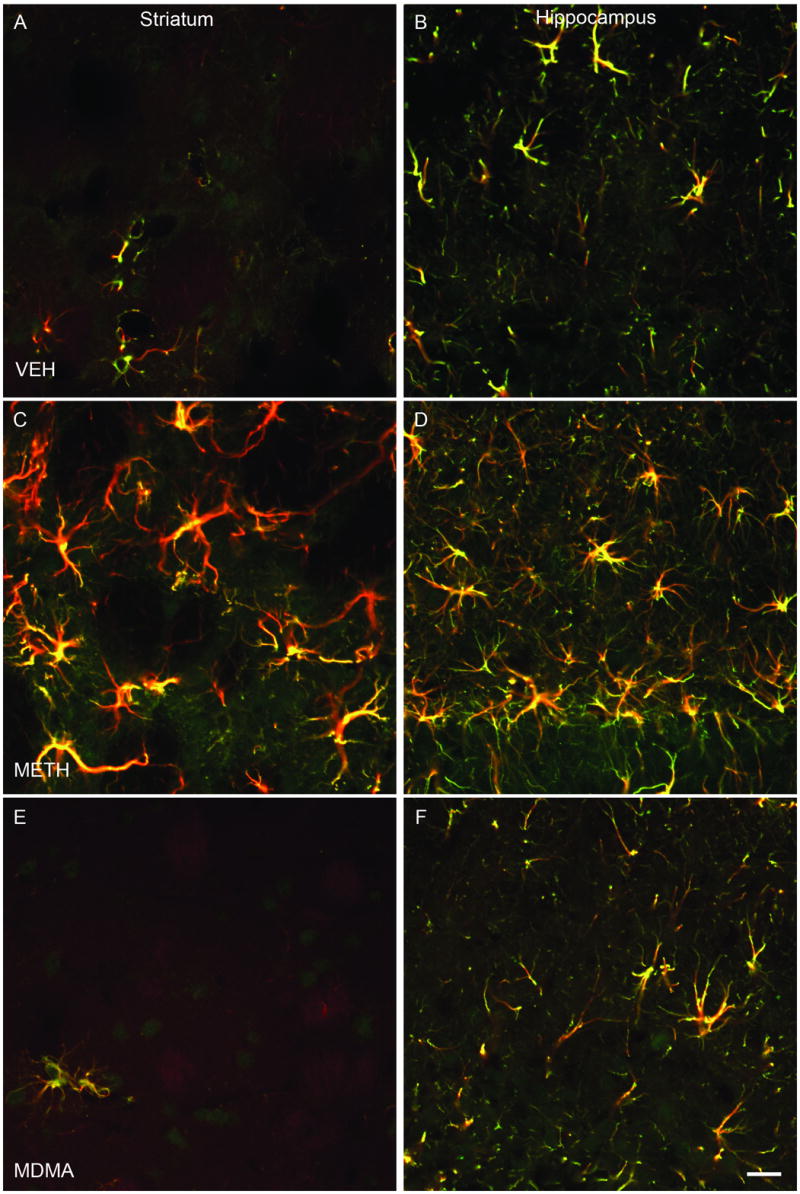
Co-localization of C-tau and GFAP immunoreactivity in striatum and dentate gyrus regions of the hippocampus. Dual fluorescent immunocytochemistry was performed with antibodies to C-tau7 and GFAP and striatal (panels A, C, E) and hippocampal (panels B, D, F) tissue from rats killed 3 days after treatment with VEH (panels A and B), METH (panels C and D) or MDMA (panels E and F) according to the regimen described in detail in the legend of Figure 1. Presence of yellow color indicates co-localization of C-tau and GFAP antibodies. Magnification, 40x.
Discussion
The key findings of this study are that dosing regimens of METH or AMPH, psychostimulant drugs demonstrated to produce significant long-term depletions of striatal 5-HT and DA (METH) or DA alone (AMPH) produced a significant increase in the number of C-tau immunoreactive cells in the striatum and the CA2/CA3 and dentate gyrus regions of the hippocampus. In contrast, the psychostimulants MDMA and PMA, and 5,7-DHT, a known 5-HT neurotoxin, which produce a selective depletion of 5-HT, did not increase the number of C-tau immunoreactive cells in either the striatum or hippocampus. Additionally, colocalization of C-tau and GFAP, as demonstrated with dual fluorescent immunocytochemistry and confocal microscopy, indicated that C-tau positive cells in the striatum and hippocampus of both VEH- and METH-treated animals were astrocytes.
It has been well documented that AMPH and METH produce selective depletions of striatal DA and/or 5-HT in the rat (Kogan et al., 1976, Ricaurte et al., 1980, Wagner et al., 1980) and that MDMA produces a selective long-term depletion of 5-HT (Schmidt, 1987, Battaglia et al., 1988). In the present study, repeated administration of PMA also resulted in a selective depletion of striatal 5-HT. To our knowledge, this is the first demonstration of PMA-induced 5-HT depletion.
Increased C-tau formation has been demonstrated following induction of experimental bacterial meningitis (Irazuzta et al., 2000, Irazuzta et al., 2001) and traumatic brain injury (Gabbita et al., 2005), as well as following the administration of the known neurotoxins kainic acid (Zemlan et al., 2003) and trimethyltin (Straiko et al., 2005 (submitted)). Additionally, Wallace et al. (2003) have reported that C-tau concentrations determined by ELISA are increased in the striatum and hippocampus following administration of a monoamine-depleting regimen of METH. The results of the present study, in which the number of C-tau immunoreactive cells in the striatum and hippocampus was increased following METH treatment, are in accord with the findings of Wallace et al (2003).
Upon quantifying C-tau immunoreactive cells, it was noted further that the C-tau positive cells exhibited a stellate morphology and distribution that was consistent with that of astrocytes. Although a similar morphology of C-tau immunoreactive cells was evident in an earlier study (Zemlan et al., 2003), dual immunohistochemistry was not employed in that study to distinguish neuronal vs. astrocytic localization of C-tau. Fluorescence immunocytochemistry and confocal analysis in the present study revealed the co-localization of C-tau and GFAP immunoreactivity. Furthermore, in a separate experiment, there was no overlap in the localization of immunoreactivity for C-tau and the neuronal marker NeuN (unpublished observations). These results further support the conclusion that C-tau positive cells were astroctyes. However, additional studies are necessary to determine whether C-tau immunoreactivity in astrocytes results either from C-tau of neuronal origin that is scavenged by astrocytes, leading to its localization within these cells or from C-tau isoforms that always reside within astrocytes. Although tau is usually associated with the axons of neurons, tau has also been shown to be present in glial cells (Bloom et al., 1984, LoPresti et al., 1995, Yoshiyama et al., 2003). Tau accumulation in glia is the result of increased synthesis or reduced clearance of the molecule that results in tau fibrillization and degeneration of the affected cells (Yoshiyama et al., 2003). Thus, it is possible that C-tau is a novel marker for reactive astrocytosis in response to CNS insult. In this regard, it is noteworthy that numerous CNS insults (e.g. kainic acid, trimethyltin, traumatic brain injury) that have been shown to increase C-tau formation (Irazuzta et al., 2001, Zemlan et al., 2003, Gabbita et al., 2005) also increase GFAP, a marker of reactive gliosis (Brock and O’Callaghan, 1987, O’Callaghan and Miller, 1993, McCann et al., 1996, Koczyk and Oderfeld-Nowak, 2000, Little et al., 2002, Wang et al., 2004a, Guo et al., 2006).
Furthermore, the results of the present experiment, in which the number of C-tau immunoreactive cells has been shown to be increased in the striatum following METH or AMPH treatment, but not MDMA or PMA treatment, are in agreement with previous studies that have examined GFAP immunoreactivity or protein expression. Treatment of rodents with a METH regimen similar to that used in this study or single high-dose METH administration has been reported to produce an increase in GFAP in the striatum as measured by semi-quantitative immunoblot (Fukumura et al., 1998) or immunoreactivity (Bowyer et al., 1994, Miller and O’Callaghan, 1994, O’Callaghan and Miller, 1994, Broening et al., 1997) 3 days after drug treatment. Various dosing regimens of AMPH also have been reported to increase striatal GFAP immunoreactivity (Bowyer, 2000, Armstrong et al., 2004, Bowyer et al., 2004). According to O’Callaghan and Miller (1993), reactive gliosis may be elicited by damage to any neuronal or glial cell type. Although most studies have focused on neuronal toxicity following administration of psychostimulant drugs, studies with primary astrocyte cultures suggest that METH also may act directly on astrocytes to produce astrogliosis and oxidative stress (Stadlin et al., 1998). Thus, the increase in C-tau in response to METH or AMPH may be reflective of damage to either neurons or astrocytes in these brain regions.
In contrast to the pronounced effect of METH and AMPH on the number of C-tau immunoreactive cells, treatment of rats with a 5-HT depleting regimen of MDMA failed to significantly increase the number of C-tau positive cells in the striatum and hippocampus; similarly, PMA was without effect on C-tau immunoreactivity in the striatum. Although only a limited number of amphetamine analogs was examined in the present study, it is intriguing that an increase in the number of C-tau immunoreactive cells was produced by psychostimulants (e.g., METH and AMPH) that produce DA depletion but not by those (e.g., MDMA, PMA) that produce selective 5-HT depletion.
The lack of C-tau response following MDMA treatment is generally consistent with the results from several studies that have assessed the effects of MDMA on GFAP. Repeated high-dose administration of MDMA that results in significant 5-HT depletion has been reported to have no effect on GFAP immunoreactivity (O’Callaghan, 2002b, Pubill et al., 2003, Wang et al., 2004b), although Aguirre et al. (1999) have reported a GFAP response to MDMA. O’Callaghan and Miller (1993) also have reported that multiple injections of exceptionally high doses of MDMA produce an increase in GFAP that is, nevertheless, relatively modest when compared to the response to METH. However, MDMA does produce a marked increase in GFAP in the mouse (O’Callaghan and Miller, 1994), but it should be noted that MDMA produces DA, not 5-HT, depletion in the mouse. This pattern of response in the mouse also appears to be consistent with the differential effects of DA and 5-HT depleting amphetamine analogs on C-tau and GFAP expression.
The lack of GFAP response and inconsistencies in silver staining following a 5-HT-depleting regimen of MDMA have led to debate as to whether MDMA is neurotoxic to 5-HT axon terminals (Commins et al., 1987, Jensen et al., 1993, O’Callaghan and Miller, 1993, Pubill et al., 2003). In the present study, the lack of an increase in the number of C-tau positive astrocytes in response to MDMA and PMA also could be viewed as indirect evidence that these amphetamine analogs are not neurotoxic. However, the i.c.v. administration of the prototypic 5-HT neurotoxin 5,7-DHT produced a 70% reduction in striatal 5-HT concentrations but did not increase the number of C-tau immunoreactive cells. Thus, it would appear that C-tau expression is not a reliable marker of degenerating 5-HT axon terminals. It could be suggested that the extent of 5-HT depletion produced by MDMA or 5,7-DHT may not have been sufficient to elicit a C-tau response. However, the magnitude of DA depletion produced by METH administration was no greater than the magnitude of 5-HT depletion produced by MDMA or 5,7-DHT, yet METH did produce an increase in C-tau positive cells. Alternatively, degenerating 5-HT axon terminals may not trigger an astrocytic response. The i.c.v. administration of 5,7-DHT not only fails to increase C-tau formation, as shown in the present study, but also produces little or no increase in GFAP message or protein expression (O’Callaghan and Miller, 1993, Rowland et al., 1993, Bendotti et al., 1994, Fages et al., 1994, Dugar et al., 1998, Wang et al., 2004b). Reasons for the failure of 5-HT neurotoxins to produce robust histological or immunocytochemical markers of toxicity are unknown.
In summary, treatment of rats with DA-depleting regimens of METH or AMPH resulted in an increase in the number of C-tau immunoreactive cells in the striatum and hippocampus. In contrast, the psychostimulants MDMA and PMA, as well as 5,7-DHT, which produce selective depletions of brain 5-HT, did not increase C-tau immunoreactivity. Furthermore, C-tau formation was localized exclusively to astrocytes. These data are consistent with previous findings that treatment with METH, but not MDMA, produces a response of reactive gliosis.
Abbreviations
- AMPH
amphetamine
- CNS
central nervous system
- C-tau
cleaved microtubule associated protein tau
- DA
dopamine
- DG
dentate gyrus
- 5
7-DHT, 5, 7-dihydroxytryptamine
- EDTA
ethylenediaminetetraacetic acid
- ELISA
enzyme-linked immunosorbant assay
- 5-HT
5-hydroxytryptamine
- GFAP
glial fibrillary acidic protein
- MAP-tau
microtubule associated protein tau
- MDA
methylenedioxyamphetamine
- MDMA
±3, 4-methylenedioxymethamphetamine
- METH
methamphetamine
- PBS
phosphate buffered saline
- PMA
para-methoxyamphetamine
- SERT
serotonin transporter
- STR
striatum
- TBS
tris buffered saline
- TBST
tris buffered saline containing 1% Tween
- VEH
vehicle
Footnotes
Section Editor: Yoland Smith
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Armstrong V, Reichel CM, Doti JF, Crawford CA, McDougall SA. Repeated amphetamine treatment causes a persistent elevation of glial fibrillary acidic protein in the caudate-putamen. Eur J Pharmacol. 2004;488:111–115. doi: 10.1016/j.ejphar.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Balfour ME, Brown JL, Yu L, Coolen LM. Potential contributions of efferents from medial prefrontal cortex to neural activation following sexual behavior in the male rat. Neuroscience. 2006;137:1259–1276. doi: 10.1016/j.neuroscience.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Yeh SY, De Souza EB. MDMA-induced neurotoxicity: parameters of degeneration and recovery of brain serotonin neurons. Pharmacol Biochem Behav. 1988;29:269–274. doi: 10.1016/0091-3057(88)90155-4. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Yeh SY, O’Hearn E, Molliver ME, Kuhar MJ, De Souza EB. 3,4-Methylenedioxymethamphetamine and 3,4-methylenedioxyamphetamine destroy serotonin terminals in rat brain: quantification of neurodegeneration by measurement of [3H]paroxetine-labeled serotonin uptake sites. J Pharmacol Exp Ther. 1987;242:911–916. [PubMed] [Google Scholar]
- Bendotti C, Baldessari S, Pende M, Tarizzo G, Miari A, Presti ML, Mennini T, Samanin R. Does GFAP mRNA and mitochondrial benzodiazepine receptor binding detect serotonergic neuronal degeneration in rat? Brain Res Bull. 1994;34:389–394. doi: 10.1016/0361-9230(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Bloom GS, Schoenfeld TA, Vallee RB. Widespread distribution of the major polypeptide component of MAP 1 (microtubule-associated protein 1) in the nervous system. J Cell Biol. 1984;98:320–330. doi: 10.1083/jcb.98.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer JF. Neuronal degeneration in the limbic system of weanling rats exposed to saline, hyperthermia or d-amphetamine. Brain Res. 2000;885:166–171. doi: 10.1016/s0006-8993(00)02925-5. [DOI] [PubMed] [Google Scholar]
- Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- Bowyer JF, Harris AJ, Delongchamp RR, Jakab RL, Miller DB, Little AR, O’Callaghan JP. Selective changes in gene expression in cortical regions sensitive to amphetamine during the neurodegenerative process. Neurotoxicology. 2004;25:555–572. doi: 10.1016/j.neuro.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Brock TO, O’Callaghan JP. Quantitative changes in the synaptic vesicle proteins synapsin I and p38 and the astrocyte-specific protein glial fibrillary acidic protein are associated with chemical-induced injury to the rat central nervous system. J Neurosci. 1987;7:931–942. doi: 10.1523/JNEUROSCI.07-04-00931.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broening HW, Pu C, Vorhees CV. Methamphetamine selectively damages dopaminergic innervation to the nucleus accumbens core while sparing the shell. Synapse. 1997;27:153–160. doi: 10.1002/(SICI)1098-2396(199710)27:2<153::AID-SYN6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Commins DL, Vosmer G, Virus RM, Woolverton WL, Schuster CR, Seiden LS. Biochemical and histological evidence that methylenedioxymethylamphetamine (MDMA) is toxic to neurons in the rat brain. J Pharmacol Exp Ther. 1987;241:338–345. [PubMed] [Google Scholar]
- Dugar A, Patanow C, O’Callaghan JP, Lakoski JM. Immunohistochemical localization and quantification of glial fibrillary acidic protein and synaptosomal-associated protein (mol. wt 25000) in the ageing hippocampus following administration of 5,7-dihydroxytryptamine. Neuroscience. 1998;85:123–133. doi: 10.1016/s0306-4522(97)00606-4. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Marshall JF. Methamphetamine neurotoxicity: dissociation of striatal dopamine terminal damage from parietal cortical cell body injury. Synapse. 1998;30:433–445. doi: 10.1002/(SICI)1098-2396(199812)30:4<433::AID-SYN10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Schmued LC, Marshall JF. Characterizing cortical neuron injury with Fluoro-Jade labeling after a neurotoxic regimen of methamphetamine. Synapse. 1998;30:329–333. doi: 10.1002/(SICI)1098-2396(199811)30:3<329::AID-SYN10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Fages C, Le Prince G, Didier-Bazes M, Rolland B, Hardin H, Tardy M. Long-term astroglial reaction to serotonergic fiber degeneration. Brain Res. 1994;639:161–166. doi: 10.1016/0006-8993(94)91777-9. [DOI] [PubMed] [Google Scholar]
- Fukumura M, Cappon GD, Pu C, Broening HW, Vorhees CV. A single dose model of methamphetamine-induced neurotoxicity in rats: effects on neostriatal monoamines and glial fibrillary acidic protein. Brain Res. 1998;806:1–7. doi: 10.1016/s0006-8993(98)00656-8. [DOI] [PubMed] [Google Scholar]
- Gabbita SP, Scheff SW, Menard RM, Roberts K, Fugaccia I, Zemlan FP. Cleaved-tau: a biomarker of neuronal damage after traumatic brain injury. J Neurotrauma. 2005;22:83–94. doi: 10.1089/neu.2005.22.83. [DOI] [PubMed] [Google Scholar]
- Gudelsky GA, Yamamoto BK, Nash JF. Potentiation of 3,4-methylenedioxymethamphetamine-induced dopamine release and serotonin neurotoxicity by 5-HT2 receptor agonists. Eur J Pharmacol. 1994;264:325–330. doi: 10.1016/0014-2999(94)90669-6. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sayeed I, Baronne LM, Hoffman SW, Guennoun R, Stein DG. Progesterone administration modulates AQP4 expression and edema after traumatic brain injury in male rats. Exp Neurol. 2006 doi: 10.1016/j.expneurol.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- Irazuzta JE, de Courten-Myers G, Zemlan FP, Bekkedal MY, Rossi J., 3rd Serum cleaved Tau protein and neurobehavioral battery of tests as markers of brain injury in experimental bacterial meningitis. Brain Res. 2001;913:95–105. doi: 10.1016/s0006-8993(01)02764-0. [DOI] [PubMed] [Google Scholar]
- Irazuzta JE, Pretzlaff R, Rowin M, Milam K, Zemlan FP, Zingarelli B. Hypothermia as an adjunctive treatment for severe bacterial meningitis. Brain Res. 2000;881:88–97. doi: 10.1016/s0006-8993(00)02894-8. [DOI] [PubMed] [Google Scholar]
- Itzhak Y, Achat-Mendes C. Methamphetamine and MDMA (ecstasy) neurotoxicity: ‘of mice and men’. IUBMB Life. 2004;56:249–255. doi: 10.1080/15216540410001727699. [DOI] [PubMed] [Google Scholar]
- Jensen KF, Olin J, Haykal-Coates N, O’Callaghan J, Miller DB, de Olmos JS. Mapping toxicant-induced nervous system damage with a cupric silver stain: a quantitative analysis of neural degeneration induced by 3,4-methylenedioxymethamphetamine. NIDA Res Monogr. 1993;136:133–149. doi: 10.1037/e495922006-008. discussion 150-134. [DOI] [PubMed] [Google Scholar]
- Koczyk D, Oderfeld-Nowak B. Long-term microglial and astroglial activation in the hippocampus of trimethyltin-intoxicated rat: stimulation of NGF and TrkA immunoreactivities in astroglia but not in microglia. Int J Dev Neurosci. 2000;18:591–606. doi: 10.1016/s0736-5748(99)00111-2. [DOI] [PubMed] [Google Scholar]
- Kogan FJ, Nichols WK, Gibb JW. Influence of methamphetamine on nigral and striatal tyrosine hydroxylase activity and on striatal dopamine levels. Eur J Pharmacol. 1976;36:363–371. doi: 10.1016/0014-2999(76)90090-x. [DOI] [PubMed] [Google Scholar]
- Little AR, Benkovic SA, Miller DB, O’Callaghan JP. Chemically induced neuronal damage and gliosis: enhanced expression of the proinflammatory chemokine, monocyte chemoattractant protein (MCP)-1, without a corresponding increase in proinflammatory cytokines(1) Neuroscience. 2002;115:307–320. doi: 10.1016/s0306-4522(02)00359-7. [DOI] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci U S A. 1995;92:10369–10373. doi: 10.1073/pnas.92.22.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyles J, Cadet JL. Methylenedioxymethamphetamine (MDMA, Ecstasy) neurotoxicity: cellular and molecular mechanisms. Brain Res Brain Res Rev. 2003;42:155–168. doi: 10.1016/s0165-0173(03)00173-5. [DOI] [PubMed] [Google Scholar]
- McCann MJ, O’Callaghan JP, Martin PM, Bertram T, Streit WJ. Differential activation of microglia and astrocytes following trimethyl tin-induced neurodegeneration. Neuroscience. 1996;72:273–281. doi: 10.1016/0306-4522(95)00526-9. [DOI] [PubMed] [Google Scholar]
- Miller DB, O’Callaghan JP. Environment-, drug- and stress-induced alterations in body temperature affect the neurotoxicity of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:752–760. [PubMed] [Google Scholar]
- Molliver ME, Berger UV, Mamounas LA, Molliver DC, O’Hearn E, Wilson MA. Neurotoxicity of MDMA and related compounds: anatomic studies. Ann N Y Acad Sci. 1990;600:649–661. doi: 10.1111/j.1749-6632.1990.tb16916.x. discussion 661-644. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP, Miller DB. Quantification of reactive gliosis as an approach to neurotoxicity assessment. NIDA Res Monogr. 1993;136:188–212. [PubMed] [Google Scholar]
- O’Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- O’Callaghan JP, Miller DB. Neurotoxic Effects if Substituted Amphetamines in Rats and Mice: Challenges to the Current Dogma. In: Massaro EJ, editor. Handbook of Neurotoxicology. Vol. 2. Humana Press, Inc; Totowa, NJ: 2002a. pp. 269–301. [Google Scholar]
- O’Callaghan JP, Miller DB. Neurotoxic Effects of Substituted Amphetamines in Rats and Mice: Challenges to the Current Dogma. In: Massaro EJ, editor. Handbook of Neurotoxicology. Vol. 2. Humana Press, Inc; Totowa, NJ: 2002b. pp. 269–301. [Google Scholar]
- Pubill D, Canudas AM, Pallas M, Camins A, Camarasa J, Escubedo E. Different glial response to methamphetamine- and methylenedioxymethamphetamine-induced neurotoxicity. Naunyn Schmiedebergs Arch Pharmacol. 2003;367:490–499. doi: 10.1007/s00210-003-0747-y. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- Rowland NE, Kalehua AN, Li BH, Semple-Rowland SL, Streit WJ. Loss of serotonin uptake sites and immunoreactivity in rat cortex after dexfenfluramine occur without parallel glial cell reactions. Brain Res. 1993;624:35–43. doi: 10.1016/0006-8993(93)90057-t. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ. Neurotoxicity of the psychedelic amphetamine, methylenedioxymethamphetamine. J Pharmacol Exp Ther. 1987;240:1–7. [PubMed] [Google Scholar]
- Schmidt CJ, Taylor VL. Depression of rat brain tryptophan hydroxylase activity following the acute administration of methylenedioxymethamphetamine. Biochem Pharmacol. 1987;36:4095–4102. doi: 10.1016/0006-2952(87)90566-1. [DOI] [PubMed] [Google Scholar]
- Schmued LC. Demonstration and localization of neuronal degeneration in the rat forebrain following a single exposure to MDMA. Brain Res. 2003;974:127–133. doi: 10.1016/s0006-8993(03)02563-0. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Bowyer JF. Methamphetamine exposure can produce neuronal degeneration in mouse hippocampal remnants. Brain Res. 1997;759:135–140. doi: 10.1016/s0006-8993(97)00173-x. [DOI] [PubMed] [Google Scholar]
- Stadlin A, Lau JW, Szeto YK. A selective regional response of cultured astrocytes to methamphetamine. Ann N Y Acad Sci. 1998;844:108–121. [PubMed] [Google Scholar]
- Stone DM, Merchant KM, Hanson GR, Gibb JW. Immediate and long-term effects of 3,4-methylenedioxymethamphetamine on serotonin pathways in brain of rat. Neuropharmacology. 1987;26:1677–1683. doi: 10.1016/0028-3908(87)90117-1. [DOI] [PubMed] [Google Scholar]
- Stone DM, Stahl DC, Hanson GR, Gibb JW. The effects of 3,4-methylenedioxymethamphetamine (MDMA) and 3,4-methylenedioxyamphetamine (MDA) on monoaminergic systems in the rat brain. Eur J Pharmacol. 1986;128:41–48. doi: 10.1016/0014-2999(86)90555-8. [DOI] [PubMed] [Google Scholar]
- Straiko MMW, Gudelsky GA, Coolen LM, Harrison R, Zemlan FP. Treatment with trimethyltin promotes the formation of cleaved tau in the rat striatum. Journal of Neuroscience Research. 2005 doi: 10.1002/jnr.21002. (submitted) [DOI] [PubMed] [Google Scholar]
- Thomas DM, Kuhn DM. MK-801 and dextromethorphan block microglial activation and protect against methamphetamine-induced neurotoxicity. Brain Res. 2005;1050:190–198. doi: 10.1016/j.brainres.2005.05.049. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- Wallace TL, Vorhees CV, Zemlan FP, Gudelsky GA. Methamphetamine enhances the cleavage of the cytoskeletal protein tau in the rat brain. Neuroscience. 2003;116:1063–1068. doi: 10.1016/s0306-4522(02)00795-9. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yu S, Simonyi A, Rottinghaus G, Sun GY, Sun AY. Resveratrol protects against neurotoxicity induced by kainic acid. Neurochem Res. 2004a;29:2105–2112. doi: 10.1007/s11064-004-6883-z. [DOI] [PubMed] [Google Scholar]
- Wang X, Baumann MH, Xu H, Rothman RB. 3,4-methylenedioxymethamphetamine (MDMA) administration to rats decreases brain tissue serotonin but not serotonin transporter protein and glial fibrillary acidic protein. Synapse. 2004b;53:240–248. doi: 10.1002/syn.20058. [DOI] [PubMed] [Google Scholar]
- Watson RE, Jr, Wiegand SJ, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–159. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- Xie T, Tong L, McLane MW, Hatzidimitriou G, Yuan J, McCann U, Ricaurte G. Loss of Serotonin Transporter Protein after MDMA and Other Ring-Substituted Amphetamines. Neuropsychopharmacology. 2006 doi: 10.1038/sj.npp.1301031. [DOI] [PubMed] [Google Scholar]
- Yoshiyama Y, Zhang B, Bruce J, Trojanowski JQ, Lee VM. Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau-induced degeneration in astrocytes. J Neurosci. 2003;23:10662–10671. doi: 10.1523/JNEUROSCI.23-33-10662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemlan FP, Jauch EC, Mulchahey JJ, Gabbita SP, Rosenberg WS, Speciale SG, Zuccarello M. C-tau biomarker of neuronal damage in severe brain injured patients: association with elevated intracranial pressure and clinical outcome. Brain Res. 2002;947:131–139. doi: 10.1016/s0006-8993(02)02920-7. [DOI] [PubMed] [Google Scholar]
- Zemlan FP, Mulchahey JJ, Gudelsky GA. Quantification and localization of kainic acid-induced neurotoxicity employing a new biomarker of cell death: cleaved microtubule-associated protein-tau (C-tau) Neuroscience. 2003;121:399–409. doi: 10.1016/s0306-4522(03)00459-7. [DOI] [PubMed] [Google Scholar]
- Zemlan FP, Rosenberg WS, Luebbe PA, Campbell TA, Dean GE, Weiner NE, Cohen JA, Rudick RA, Woo D. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem. 1999;72:741–750. doi: 10.1046/j.1471-4159.1999.0720741.x. [DOI] [PubMed] [Google Scholar]