

Abstract
The accumulation of mildly deleterious mutations accompanying recurrent regeneration of plant germ plasm was modeled under regeneration conditions characterized by different amounts of selection and genetic drift. Under some regeneration conditions (sample sizes ≥75 individuals and bulk harvesting of seed) mutation accumulation was negligible, but under others (sample sizes <75 individuals or equalization of seed production by individual plants) mutation numbers per genome increased significantly during 25–50 cycles of regeneration. When mutations also are assumed to occur (at elevated rates) during seed storage, significant mutation accumulation and fitness decline occurred in 10 or fewer cycles of regeneration regardless of the regeneration conditions. Calculations also were performed to determine the numbers of deleterious mutations introduced and remaining in the genome of an existing variety after hybridization with a genetic resource and subsequent backcrossing. The results suggest that mutation accumulation has the potential to reduce the viability of materials held in germ plasm collections and to offset gains expected by the introduction of particular genes of interest from genetic resources.
Keywords: genetic load, germ plasm, conservation genetics, linkage drag
Collections of plant and animal germ plasm provide valuable reservoirs of biological materials for use in controlled experiments, for the selective improvement of domesticated species, and as sources of potentially new foods or drugs (1). In the case of plants, the majority of managed germ plasm resources are housed in seed banks. Historically such collections have been associated with agricultural applications, but there is increasing interest in adopting ex situ methods of conservation as part of the overall strategy for the conservation of many noneconomically important species (2).
The maintenance of seed viability presents a number of problems for the long-term conservation of plant germ plasm. Although seed preservation technology has been improved in recent years, all stored seeds eventually lose viability. Typically, when germination percentages fall below set limits (65–85% depending on the institution), regeneration of the collection from a finite sample of the stored germ plasm is recommended (1). The frequency with which regeneration must be conducted depends on the type of seeds in question. Plants with so-called “orthodox” seed, that withstand drying and subfreezing temperatures may, in many instances, be stored for decades. Cryogenic preservation in liquid nitrogen (used for a relatively small fraction of the world’s germ plasm resources) can extend storage life for 100 years or more for some species. The seeds of some plants, especially many tropical species, however, cannot tolerate drying and cold storage, and regeneration must be conducted frequently. Moreover, in many developing countries, reliable cold storage facilities are not widely available, and seed banks in these countries often rely more heavily on regeneration (and less on storage) for the maintenance of viable germ plasm.
Regeneration of stored seed renders germ plasm collections prone to loss of diversity arising from sampling error (or genetic drift) accompanying the finite size of the adult population used to regenerate the new batch of seed (3, 4). The problem of restricted sample sizes during regeneration is aggravated by pressures imposed on seed banks to manage an ever increasing number of collections (or accessions) with limited funds (5); seed banks often use the smallest number of plants possible for seed regeneration, typically fewer than 100 plants (6). Several practices have been suggested to counteract the genetic changes to the germ plasm collections that may occur during regeneration. These practices include the collection of equal aliquots of seed per plant and the use of optimal growing conditions. Such procedures should maintain effective population size close to its maximum for the number of plants used in regeneration, and minimize selective changes that otherwise might occur in response to the storage and regeneration environment (1).
A potential, but largely overlooked, problem accompanying the ex situ storage and regeneration of genetic resources is the accumulation of mildly deleterious mutations. Paradoxically, the same practices that prevent the loss of variation in response to the storage and regeneration environment are also those that allow the number of mildly deleterious mutations to build up in the germ plasm collection. Although mutation accumulation has received attention in relation to mating system evolution and the long-term viability of natural populations (7–12), there have been no detailed analyses of the problem in the context of ex situ genetic resource conservation, though studies by Couvet and Ronfort (13) and Lange (14) suggest that selection against mild deleterious mutations is weak when fertility variation among individuals is minimized.
Accumulation of mildly deleterious mutations may lead to loss of viability in germ plasm collections and, in the case of wild species, present obstacles to the use of such seed in any future efforts to reintroduce such materials into natural populations. Mutation accumulation also may complicate the use of such seed in selective breeding. In this paper we use deterministic calculations and simulations to examine how finite sample size and other storage and regeneration conditions may contribute to the accumulation of mildly deleterious mutations in germ plasm collections of self-fertilizing and outcrossing plant species, and we examine some of the consequences of mutation accumulation for the future use of materials stored in ex situ collections. In so doing, we develop a deterministic method for examining mutation accumulation in finite populations of self-fertilizing species.
THEORETICAL ANALYSES AND RESULTS
Mutation Parameters.
By observing fitness decline in laboratory populations of Drosophila melanogaster raised under conditions that minimize the opportunity for natural selection, and by using lines containing balancer chromosomes that prevent recombination (i.e., mutation accumulation experiments), Mukai and colleagues (15–17) obtained estimates of mutation rates to mildly deleterious alleles of 0.5–1.0 per genome per generation, and mean selection coefficients of 0.05–0.10 for homozygous mutations. Measures of fitness in progeny from crosses between mutation accumulation lines yielded estimates of dominance coefficients of ca. 0.35 (i.e., partial dominance) (15–18). Subsequently, inbreeding depression measured in predominantly self-fertilizing, together with crosses between inbred lines, have suggested values of genomic mutation rates and dominance coefficients for deleterious mutations that are in general agreement with those obtained in Mukai’s mutation accumulation experiments (19–21). For the majority of the work presented here, we assumed that mutations occurred only during active plant growth (i.e., at the time of regeneration). Parameter values as estimated by Mukai for Drosophila were assumed in most of the work described below. We did not investigate the accumulation of lethal mutations, as lethals are expected to be purged from the collection. Nor did we consider beneficial mutations. Although genetic changes sometimes may arise in collections and provide raw material for improvement in breeding (22), experimental evidence indicates that the majority of mutations have deleterious effects (23).
Recently there have been questions raised as to the generality of mutation rate estimates obtained in the earlier mutation accumulation experiments. For instance, mutation accumulation experiments with the nematode Caenorhabditis elegans have revealed lower mutation rate estimates (24), but mutation rate estimation is fraught with statistical difficulties (e.g., confounding of estimates of U and s; ref. 23), and uncertainty remains about the actual rates of mildly deleterious mutation, and whether these rates vary significantly between species.
Effects of Seed Aging on Mutation Rate.
Less is known about the effects of long-term seed storage on mutation rates. Several studies have suggested that loss of viability accompanying seed aging is associated with increases in the frequency of chromosomal aberrations, DNA lesions, chlorophyll deficiency mutations, as well as decreased activity levels of DNA polymerase (25–28). In some studies, small viability decreases in aged seed samples have been associated with 5-fold increases in the numbers of mutations detected at certain gene loci (27, 29). Whether and by how much overall genomic mutation rates are influenced by seed aging remains uncertain, but given the variety of evidence for genetic degradation accompanying seed aging, we thought it important to consider the possibility of elevated rates of genomic mutation during seed storage. Thus, in addition to assuming mutation rates of U = 0.5–1.0, we carried out a series of calculations of mutation accumulation in germ plasm collection assuming higher overall rates of genomic mutation, caused by mutations occurring during regeneration as well as during storage.
Regeneration.
The term “regeneration cycle” is used below to refer to the sequence of events involving seed storage, planting of a finite sample of stored seeds, and harvesting of the regenerated seeds to form the new seed collection. Mutation accumulation over 50 regeneration cycles was considered in this investigation. The specific time duration to which this number of cycles corresponds depends on the species and seed bank in question. Although in principle the conservation of germ plasm should be viewed as an ongoing endeavor, we felt it difficult to justify the use of larger numbers of regeneration cycles in our work, as evolving technologies eventually may extend seed storage periods for the majority of germ plasm collections.
Two commonly used methods of harvesting the seed used to regenerate germ plasm collections were considered. These are referred to below as the “bulk” and “equal” methods. In the bulk method, all seeds are harvested and stored in a common pool from which seeds are drawn at random to start the next regeneration cycle. Thus, if adult plants differ in their contribution to the seed pool as a result of variation in mutation load, selection will act to purge some of the deleterious mutations from the collection. The effectiveness of purging will depend on the sample size of adult plants used—i.e., small samples are more prone to fixation of mildly deleterious mutations. In the equal harvesting method, an identical aliquot of seed is collected per adult plant. Differences in mutation load among adults do not influence numerical contributions to the seed pool, and accordingly, purging of mildly deleterious is not expected. Purging of lethal mutations will, however, occur because such mutations cause seed abortion.
Sample sizes used to regenerate germ plasm collections typically vary between 25 and 100 plants per accession, a number that has been accepted in most regeneration efforts as a compromise between the need to limit the loss of variation caused by genetic drift and the demands posed by regeneration of many hundreds to thousands of accessions (6). Hence, we examined mutation accumulation with samples of 25, 50, 75, and 100 plants.
Mutation Accumulation in Cross-Fertilizing Species.
Two sources of mutations, those originally present in the infinite-sized ancestral population that was sampled to construct the initial collection (hereafter, ancestral mutations), and those arising during regeneration (new mutations) were considered. Mutations were assumed to be unlinked. Charlesworth et al. (30) have shown that linkage has little influence on mutation accumulation unless mutated loci are tightly linked (<0.1 centiMorgans). Mutation frequencies in the ancestral population were assumed to reflect mutation-selection balance. In the initial sample of K diploid individuals taken from the ancestral population, there are 2K+1 possible states for each mutable locus, ranging from loss (0 copies) to fixation (2K copies) of the mutant allele. The sample size was assumed to remain constant throughout the 50 regeneration cycles. Following Lynch et al. (12), let xa,t denote a column vector containing elements x(i)a,t=0 (i = 0, 1, … , 2K), these elements being the expected numbers of loci in each of the 2K+1 states with respect to the ancestral mutations (hence, the subscript a) at time t = 0. In the initial sample, values of x(i)a,t=0 follow a Poisson distribution; i.e., x(i)a,t=0 = exp(−2Kq)(2Kq)iM/i! where q = u/hs is the frequency of the mutant allele in the ancestral population, u is genic mutation rate, h is the dominance coefficient for deleterious mutations, s is the reduction in fitness when the mutation is homozygous (31), and M is the number of mutable loci. The corresponding genomic mutation rate is U = 2uM. Selection and dominance coefficients for deleterious mutations were assumed to be identical among loci, and fitness was assumed to be the product of multiplicative interactions among loci.
After sampling the ancestral population, the loss and fixation of ancestral mutations were assumed to be governed by drift and selection. Thus the probability that a sample in state xa,t=0 is in the new state xa,t=1 after one cycle of regeneration is given by xa,t=1 = Axa,t=0, where A is the (2K + 1) × (2K + 1) matrix of transition probabilities calculated under the Wright-Fisher model (see equation 9 in ref. 12). Under equal seed harvesting, it was assumed that s = 0 in this equation. Iteration of this system of equations T times gives the expected numbers of loci in each of the 2K+1 states after T regeneration cycles. The expected contribution of the ancestral mutations to mutation accumulation in the collection at generation T is:
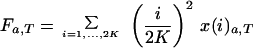 |
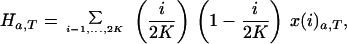 |
1 |
where Fa,T and Ha,T are numbers of homozygous and heterozygous mutations at time T and i/2K and (1 − i/2K) are frequencies of mutant and wild-type alleles at single loci.
For new mutations, let xn,t denote a column vector with elements x(i)n,t (i = 0, 1, … , 2K), the expected numbers of loci in each of the 2K+1 states with respect to new mutations at time t. This vector initially (before regeneration begins) contains all zero elements. During each regeneration cycle there are U new mutations produced per genome. Accordingly, the element x(1)n,t is incremented by the product UK during each cycle, starting at t = 1. The values of the elements in the vector xn,t change because of selection and drift during recurrent regeneration, as governed by the probabilities in matrix A. The number of new homozygous (Fn,t) and heterozygous (Hn,t) mutations at generation T were calculated as in Eq. 1 replacing x(i)a,T by x(i)n,T. The total numbers of homozygous and heterozygous mutations in the collection then were determined by taking the sums Fa,T + Fn,T and Ha,T + Hn,T, respectively. For bulk seed harvesting, where some selection against deleterious mutations is expected to occur, the methods outlined above are conservative, as they do not take into account any selective interference among loci (12).
An independent check on the deterministic results was obtained by Monte Carlo simulation as described in Charlesworth et al. (32). Thus, as a starting point, the procedure described by Kondrashov (33) for calculating the expected frequencies (qx,y) of individuals with x heterozygote and y homozygote mutations in the ancestral population (with genomic mutation rate U) was used. The initial collection was simulated by randomly drawing a sample of K individuals from the distribution qx,y. To simulate regeneration, a sample of K progeny subsequently was drawn at random from the initial sample, and mating was simulated. Under the bulk method, a genotype’s probability of contributing to the pollen and seed pool was equal to its frequency weighted by its fitness (determined by mutation load), whereas under the equal method, the probabilities of pollen and seed donations were independent of mutation load. Fitness was assumed to be determined by multiplicative interactions among the mutated loci, wxy = (1 − hs)x(1 − s)y. U new mutations per individual were assumed to arise each generation (at loci not already bearing mutations). To simulate subsequent cycles of regeneration, sampling and mating were carried out again as above. For both simulated methods of regeneration (bulk and equal) we recorded the average number of heterozygous and homozygous mutations per individual and calculated the population average.
Fig. 1 illustrates trajectories of mutation accumulation and resulting fitness decline for simulations and deterministic analyses when U = 1.0, h = 0.35, and s = 0.05 for samples of varying size K. There was generally good correspondence between trajectories obtained by deterministic calculations and simulation (i.e., regression of simulated numbers of heterozygous and homozygous mutations at time t on those calculated deterministically gave y-intercepts and slopes near 0 and 1, respectively, with r2 > .95), and so simulation results are shown only for K = 25. In the case of bulk seed harvesting, homozygous mutations increased in number, accompanied by loss of mutations in the heterozygous state (Fig. 1A). Mutation accumulation and fitness decline were negligible for K = 75 and 100. For K = 25 and 50 the number of homozygous mutations increases roughly 10-fold and 5-fold, respectively, and fitness declines by up to 25% relative to that of the initial collection (Fig. 1C). In the case of equal seed harvesting, there is an up to 20-fold increase in numbers of homozygous mutations, and fitness declines by 50–70% over the 50 regeneration cycles, depending on the value of K (Fig. 1C).
Figure 1.

Mutation accumulation and fitness decline accompanying recurrent regeneration of germ plasm in an outcrossing plant species, with U = 1, h = 0.35, and s = 0.05. (A) Under bulk seed harvesting. Solid lines show results for deterministic analyses; dashed lines for simulation (K = 25 only). (B) Under equal seed harvesting. Dashed and solid lines as above. (C) Fitness decline relative to initial collection. Solid and dashed lines indicate bulk and equal harvesting, respectively.
Mutation Accumulation in Self-Fertilizing Species.
Allelic states at each locus are correlated under self-fertilization, and so we characterized the sample in terms of genotypic rather than allelic states. Denoting the wild-type and mutant alleles at a given locus as A1 and A2, respectively, and letting D, H, and R be the number of A1A1, A1A2, and A2A2 genotypes in the sample, we note that a sample of size K may exist in any of (K + 1)(K + 2)/2 states ranging from loss (D = K, H = 0, R = 0) to fixation (D = 0, H = 0, R = K) of the mutant allele.
For mutations inherited from the ancestral population, let ga,t denote a column vector containing elements that are the expected number of loci in each of the (K+1)(K+2)/2 possible genotypic states at time t. To obtain the initial condition, ga,t=0 we assume an infinite population in mutation-selection equilibrium is sampled. The values of ga,t=0 were determined from multinomially sampling probabilities based on expected frequencies of genotypes heterozygous and homozygous for mutations at individual loci (which are functions of u, h, and s), as derived by Ohta and Cockerham (34), and multiplied by the number of mutable loci M. As above, U = 2uM.
Next we calculated the changes in genotypic composition of the sample over time (caused by selection and drift) for mutations inherited from the ancestral population, and then for new mutations. The probability that the sample in the next generation has a particular genotypic composition (Dt+1, Ht+1, Rt+1) given the genotypic composition of the present sample (Dt, Ht, Rt) is:
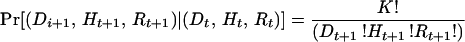 |
2 |
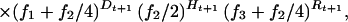 |
where f1 = (Dt/K)(1/wt), f2 = (Ht/K)[(1−hs)/wt], and f3 = (Rt/K)[(1−s)/wt] are genotype frequencies weighted by relative fitnesses, and wt = [Dt + Ht(1−hs) + Rt(1−s)]/K is the average fitness of the sample at time t. For the equal seed harvesting method, s = 0. The probabilities in Eq. 2 form the elements of a [(K+1)(K+2)/2] × [(K+1)(K+2)/2] matrix denoted P. The expected number of loci in each of the genotypic states (for mutations from the ancestral population) at time t = 1 is:
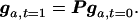 |
3 |
Iteration of this equation T times gives the state of the sample after T regeneration cycles.
For new mutations, let gn,t denote a column vector with elements that are the expected numbers of loci in each of the (K + 1)(K + 2)/2 possible genotypic states at time t. New mutations were assumed to arise in a heterozygous state, and so the single element of gn,t indexed by the genotypic combination (D = 0, H = K, R = 0) was incremented by the value UK each generation. The transition Eq. 3 again was used to calculate the genotypic state of the population in subsequent regeneration cycles. Contributions from the vectors ga,t and gn,t are summed over mutable loci to determine total numbers of heterozygous and homozygous mutations. To simulate mutation accumulation during recurrent regeneration in finite samples of self-fertilizing plants we used procedures similar to those described above for outcrossers.
Fig. 2 illustrates trajectories of mutation accumulation and fitness decline obtained from simulation and deterministic analysis when U = 1.0, h = 0.35, and s = 0.05. Regression of simulation results on those of deterministic calculations again indicates a close fit (results from simulations not shown). Because under selfing, alleles rapidly become homozygous, the number of heterozygous mutations are not shown. With bulk seed harvesting, with K ≤ 50 after 50 regeneration cycles, there is a 50–100% increase in the number of homozygous mutations, and consequent fitness reduction of 20–60%, whereas for K ≥ 75, mutation numbers increase by <50% and fitness decline is less pronounced (Fig. 2 A and C). With equal seed harvesting, mutation numbers increase nearly 4-fold and fitness declines by 75% over 50 regeneration cycles (Fig. 2 B and C). There is little effect of sample size on these trajectories.
Figure 2.

Mutation accumulation during recurrent regeneration of a self-fertilizing plant species. See Fig. 1 legend for details (simulation results not shown).
Mutation Accumulation Under Elevated Mutation Rates.
On the basis of the available information about seed aging and its effects on mutation, we considered the possibility that mutation accumulation in germ plasm samples arises not only from mutations that occur during regeneration, but from those occurring during storage (and at a higher rate, as suggested from studies referenced above). Extrapolating from the observed increases in mutation rates at single loci in artificially aged seed (27, 29), the mutation accumulation calculations for outcrossing and self-fertilizing plants were repeated, assuming U = 5 deleterious mutations per genome per generation (h = 0.35, s = 0.05, as before). In outcrossers, under bulk and equal seed harvesting, both heterozygous and homozygous mutation accumulation occurs at a more rapid rate, and there is a fitness decline of 50% or more (with equal harvesting) within the first 10 regeneration cycles (Fig. 3 A and B). In selfers, the rate of homozygous mutation accumulation and fitness decline also are increased by roughly 5-fold (Fig. 3 C and D).
Figure 3.

Mutation accumulation and fitness decline accompanying recurrent regeneration of germ plasm, with U = 5, h = 0.35, and s = 0.05. K = 50 parents for all results shown. (A) Accumulation of heterozygous and homozygous mutations in an outcrossing species. (B) Accumulation of homozygous mutations in a self-fertilizing species. (C) Fitness decline relative to initial collection in an outcrossing species. (D) Fitness decline relative to initial collection in a self-fertilizing species. Solid and dashed lines indicate bulk and equal harvesting, respectively.
Mutation Accumulation and Backcross Breeding.
One of the ways by which particular genes of interest from genetic resources are introduced into existing crop varieties is by hybridization and subsequent backcrossing using the existing variety as recurrent parent. For example, such methods often are used for introducing disease-resistance genes from less well-adapted sources into existing varieties. Backcrossing for five or six generations together with selection for the donor trait and phenotype of the existing variety (35) leads to near-isogenic lines of the original variety containing the trait of interest from the donor. Under selection for the recurrent parent phenotype, most genes on unlinked segments of the donor chromosome are lost quickly (36). There will, however, remain in the final product a residual portion of donor genome that is linked to the selected donor gene(s), and this portion will decay more slowly (as a function of the recombination rate). This phenomenon is known as “linkage drag” (36). We calculated the expected numbers of deleterious mutations introduced from the donor and remaining in the backcross-bred product when the donor genome initially contains Q accumulated mutations in a homozygous state. For brevity, we consider in detail only the case of autogamous donor and recipient. The number of donor mutations remaining in the backcross-bred material will depend on the number of donor genes selected, the number of generations of backcrossing, the chromosomal positions of the donor gene(s) with respect to deleterious mutations, and the recombination rate in the vicinity of the donor genes. For simplicity, we assumed that deleterious mutations are randomly distributed among the donor chromosomes, and that recombination rate is constant throughout the genome.
Consider an autogamous donor and existing variety, or an outcrossing species in which inbred lines for hybrid seed production are developed. By using Stam’s method (36) for analyzing linkage drag with a single donor gene, the expected length (in centiMorgans) of the linked donor chromosome remaining after B generations of backcrossing is E(xlinked, B, L, p) = (1/B)[(1 − e−Bp) + (1 − e−B(L−p))], where L denotes the length of the chromosome and p denotes the proportional position of the selected gene with respect to the end of its chromosome. This expectation derives from the probability of no crossing-over in the linked chromosome segment, multiplied by the total length of the segment. Genome segments from the donor arising from chromosomes unlinked to the selected gene(s) decay more rapidly (37), with expected residual length per chromosome of E(xunlinked, B, L) = L(1/2)B. The vast majority of residual introgressed donor genome derives from the segments linked to the selected donor gene(s). When there are C (haploid number) chromosomes each of length L, and when d different genes (d ≥ 1) are selected from the donor (with each selected donor gene on a separate chromosome, at position pd), the expected length of donor genome remaining after B generations of backcrossing is given by the sum:
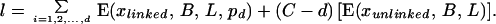 |
4 |
After B generations of backcross-breeding there is an expected number of Q(l/CL) donor mutations remaining in the recipient line.
Expected numbers of mutations remaining in a backcross-bred variety were calculated for haploid genomes of sizes of 1,000 and 3,000 centiMorgans with several different combinations of chromosome number and length (Table 1). These genome sizes are near those estimated for several major crop species. Considering a gene donor with Q = 40 homozygous mutations per genome (e.g., as expected under some of the regeneration conditions considered above), and five generations of backcross-breeding (plus one generation of selfing), one or two deleterious mutations are likely to remain in the final breeding product. For a donor with initially Q = 20 homozygous mutations per genome, these numbers are halved (data not shown). Additional generations of backcrossing, beyond the numbers normally employed by breeders (e.g., 7–9 or more generations), reduce the expected number of retained mutations to less than 1.0 (Table 1).
Table 1.
Expected numbers of homozygous mutations remaining in autogamous lines hybridized to an autogamous donor species containing Q = 40 homozygous mutations and backcrossed for B generations
| Genome size in centiMorgans, haploid number of chromosomes | Number of backcross generations (B) | Expected number of mutations remaining in a backcross-bred variety when d donor genes are selected*
|
||
|---|---|---|---|---|
| d = 1 | d = 2 | d = 3 | ||
| 1,000; 5 | 5 | 1.8 | 3.0 | 4.2 |
| 1,000; 5 | 7 | 1.1 | 2.1 | 3.1 |
| 1,000; 5 | 9 | 0.8 | 1.6 | 2.4 |
| 3,000; 5 | 5 | 0.9 | 1.2 | 1.6 |
| 3,000; 5 | 7 | 0.5 | 0.8 | 1.1 |
| 3,000; 5 | 9 | 0.3 | 0.6 | 0.8 |
| 3,000; 10 | 5 | 1.0 | 1.4 | 1.8 |
| 3,000; 10 | 7 | 0.5 | 0.8 | 1.1 |
| 3,000; 10 | 9 | 0.3 | 0.6 | 0.8 |
Numbers in this table should be halved for Q = 20 mutations.
After B generations of backcrossing and one generation of self-fertilization.
DISCUSSION
Recurrent regeneration may diminish the value of germ plasm collections, because alleles are lost during each regeneration cycle caused by genetic drift or selection (3, 4). The existence of these problems is now widely acknowledged, and recommendations have been made to ameliorate them (e.g., regeneration of plants under optimal conditions for growth, using sufficiently large sample sizes, and controlling of fecundity variation during regeneration) (6). On the other hand, consideration of the risks posed by the accumulation of deleterious mutation is largely missing from discussions of germ plasm regeneration.
The results above have shown that as recurrent regeneration progresses, there may be loss of fitness among plants in the collection because of mutation. From the standpoint of conservation, this loss could create obstacles to the future use of such materials in efforts to enhance the diversity of small populations, or in using the stored germ plasm as a source for reintroduction of the species in question into natural habitats, two potentially important reasons for conducting large-scale ex situ conservation of significant fractions of the world’s nondomesticated germ plasm, as already initiated in projects such as the Millennium Seed Bank at Kew Gardens in the United Kingdom (2, 38). Mutation accumulation in germ plasm collections also may lead to problems when attempting to use such material in plant improvement; e.g., viable material may be difficult to obtain because of poor germination, growth, and reproductive failure associated with mutation accumulation. Moreover, as the number of regeneration cycles increases, mutations may build up in the stored germ plasm to the point where one or more deleterious mutations are likely to be introduced whenever traits are introgressed from the donor genetic resource into the recipient variety. The severity of these problems is dependent on deleterious mutation rates during growth and seed storage. A general overview is difficult to provide. The estimation of mutation rates, as well as of the dominance and selection coefficients associated with deleterious mutation is an active area of research (24, 39–41), but data are still inadequate to reject or accept with confidence the values assumed above, especially in the case of mutations occurring during seed storage. Also unknown is the efficacy of selection against deleterious mutations during regeneration. If the selective growth environment used during regeneration is mild (42), there may be little expression of deleterious mutations, allowing significant mutational build up in the collection even where bulk seed harvesting and larger regeneration samples are used. In the case of germ plasm used for selective breeding, the severity of the problem is partly dependent on the expected benefits of introgressing genes from genetic resources relative to the expected deleterious effects of linked mutations. Yield increases of 8–10% accompanying backcross breeding often are considered normal, whereas increases of 15% or more are considered exceptional (43). Thus, if one or two deleterious mutations remain after backcrossing and are fully expressed (with s = .05 as assumed above), negative effects on yield of ca. 5–10% could be expected to accompany backcross-breeding, thereby largely negating increases in yield expected from the incorporation of donor traits. Additional backcross generations may be required (i.e., to “cleanse” the genome of the backcross-bred material of deleterious mutations), thus adding costs to the use of the germ plasm. Alternatively, molecular marker-assisted methods, particularly those using flanking markers to precisely isolate the genes of interest from the donor genome (44) may be helpful in species where sufficiently detailed genetic maps exist (45).
Another potential consequence of mutation accumulation in germ plasm conservation, particularly in inbreeding species, is the expected loss, caused by background selection, of neutral and near-neutral genetic variation at loci that are linked to deleterious mutations. This process has been shown to significantly reduce neutral variation when considered in theoretical analyses of self-fertilizing population (46).
Though loss of allelic variation caused by drift remains the primary conservation genetics problem accompanying regeneration of ex situ conserved germ plasm, our results suggest that in the longer term, the accumulation of deleterious mutations cannot be ignored. More detailed studies of mutation rates in natural populations and in germ plasm collections are needed to help evaluate the potential severity of the problem.
Acknowledgments
We thank Dr. Tony Brown for encouraging us to pursue this study, Drs. Deborah Charlesworth, Denis Couvet, Martin Morgan, Jean-Marie Prosperi, and Joelle Ronfort for discussions of the work presented here, Dr. Peter Keightley for sharing his unpublished manuscript with us, and two anonymous reviewers for their comments on an earlier version of the paper. D.J.S. acknowledges support from the Natural Sciences and Engineering Research Council of Canada and from the Institut National de la Recherche Agronomique (France).
References
- 1.Frankel O H, Brown A H D, Burdon J J. The Conservation of Plant Biodiversity. Cambridge, U.K.: Cambridge Univ. Press; 1995. [Google Scholar]
- 2.Smith R D, Linington S H. In: Plants for Food and Medicine. Etkin N L, Harris D R, Prendergast H D V, editors. Kew, U.K.: Royal Botanical Gardens; 1997. , in press. [Google Scholar]
- 3.Crossa J, Vencovsky R. Theor Appl Genet. 1994;89:936–942. doi: 10.1007/BF00224521. [DOI] [PubMed] [Google Scholar]
- 4.Brown A H D, Brubaker C L, Grace J P. Crop Sci. 1997;37:7–13. [Google Scholar]
- 5.Clark R L, Shands H L, Bretting P K, Eberhart S A. Crop Sci. 1997;37:1–6. [Google Scholar]
- 6.Breese E L. International Board for Plant Genetic Resources. Rome: FAO; 1989. [Google Scholar]
- 7.Charlesworth B. Genet Res. 1990;55:199–221. doi: 10.1017/s0016672300025532. [DOI] [PubMed] [Google Scholar]
- 8.Charlesworth D, Morgan M T, Charlesworth B. Evolution. 1990;44:1469–1489. doi: 10.1111/j.1558-5646.1990.tb03839.x. [DOI] [PubMed] [Google Scholar]
- 9.Kondrashov A S. Genet Res. 1984;44:199–217. doi: 10.1017/s0016672300026392. [DOI] [PubMed] [Google Scholar]
- 10.Crow J F. Oxf Surv Evol Biol. 1993;9:3–42. [Google Scholar]
- 11.Lande R. Evolution. 1994;48:1460–1469. doi: 10.1111/j.1558-5646.1994.tb02188.x. [DOI] [PubMed] [Google Scholar]
- 12.Lynch M, Conery J, Burger R. Am Nat. 1995;146:489–518. [Google Scholar]
- 13.Couvet D, Ronfort J. In: Conservation Genetics. Loeschcke V, Tomiuk J, Jain S K, editors. Basel: Birkhauser; 1994. pp. 55–68. [Google Scholar]
- 14.Lange K. Math Biosci. 1981;54:71–78. [Google Scholar]
- 15.Mukai T, Yamazaki T. Genetics. 1968;59:513–535. doi: 10.1093/genetics/59.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukai T. Genetics. 1969;61:749–761. doi: 10.1093/genetics/61.3.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohnishi O. Genetics. 1977;87:529–545. doi: 10.1093/genetics/87.3.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simmons M J, Crow J F. Annu Rev Genet. 1977;11:49–78. doi: 10.1146/annurev.ge.11.120177.000405. [DOI] [PubMed] [Google Scholar]
- 19.Charlesworth B, Charlesworth D, Morgan M T. Nature (London) 1990;347:380–382. [Google Scholar]
- 20.Charlesworth D, Lyons E, Litchfield L. Proc R Soc London Ser B. 1994;258:209–214. [Google Scholar]
- 21.Johnston M, Schoen D J. Science. 1995;267:226–229. doi: 10.1126/science.267.5195.226. [DOI] [PubMed] [Google Scholar]
- 22.Rasmusson D C, Phillips R L. Crop Sci. 1997;37:303–310. [Google Scholar]
- 23.Keightley, P. D. & Ohnishi, O. (1998) Genetics, in press. [DOI] [PMC free article] [PubMed]
- 24.Keightley P D, Caballero A. Proc Natl Acad Sci USA. 1997;94:3823–3827. doi: 10.1073/pnas.94.8.3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murata M, Tsuchiya T, Roos E. Bot Gaz. 1982;143:111–116. [Google Scholar]
- 26.Cheah K S E, Osbourne D J. Nature (London) 1978;272:593–599. doi: 10.1038/272593a0. [DOI] [PubMed] [Google Scholar]
- 27.Floris C, Meletti P. Mut Res. 1972;14:118–122. doi: 10.1016/0027-5107(72)90114-5. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi H, Naito T, Tatara A. Jpn J Genet. 1978;53:133–135. [Google Scholar]
- 29.Dourado A M, Roberts E H. Ann Bot. 1984;54:781–790. [Google Scholar]
- 30.Charlesworth D, Morgan M T, Charlesworth B. Genet Res. 1992;59:49–61. doi: 10.1017/s0016672300030160. [DOI] [PubMed] [Google Scholar]
- 31.Haldane J B S. Am Nat. 1937;71:337–349. [Google Scholar]
- 32.Charlesworth D, Morgan M T, Charlesworth B. Genet Res. 1993;61:39–56. [Google Scholar]
- 33.Kondrashov A S. Genetics. 1985;111:635–653. doi: 10.1093/genetics/111.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohta T, Cockerham C C. Genet Res. 1974;23:191–200. doi: 10.1017/s0016672300014816. [DOI] [PubMed] [Google Scholar]
- 35.Allard R W. Principles of Plant Breeding. New York: Wiley; 1960. [Google Scholar]
- 36.Stam P, Zeven A C. Euphytica. 1981;30:227–238. [Google Scholar]
- 37.Bartlett M S, Haldane J B S. J Genet. 1935;31:327–340. [Google Scholar]
- 38.Wildt D E, Seal U S, Rall W F. In: Genetic Conservation of Salmonoid Fishes. Cloud J G, Thorgaard G H, editors. New York: Plenum; 1993. pp. 159–172. [Google Scholar]
- 39.Houle D, Hoffmaster D K, Assimacopoulos S, Charlesworth B. Nature (London) 1992;359:717–719. doi: 10.1038/359058a0. [DOI] [PubMed] [Google Scholar]
- 40.Fu Y-B, Ritland K. Genetics. 1994;136:323–331. doi: 10.1093/genetics/136.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kibota T T, Lynch M. Nature (London) 1996;381:694–696. doi: 10.1038/381694a0. [DOI] [PubMed] [Google Scholar]
- 42.Kondrashov A, Houle D. Proc R Soc London Ser B. 1994;258:221–227. doi: 10.1098/rspb.1994.0166. [DOI] [PubMed] [Google Scholar]
- 43.Jinhua X, Grandillo S, Ahn S N, McCouch S R, Tanksley S D, Li J, Yuan L. Nature (London) 1996;384:224–225. [Google Scholar]
- 44.Visscher P M, Haley C S, Thompson R. Genetics. 1996;144:1923–1932. doi: 10.1093/genetics/144.4.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanksley S D, McCouch S R. Science. 1997;277:1063–1066. doi: 10.1126/science.277.5329.1063. [DOI] [PubMed] [Google Scholar]
- 46.Charlesworth B, Morgan M T, Charlesworth D. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]