

Abstract
The essential pre-mRNA splicing factor, U2 Auxiliary Factor 65KD (U2AF65) recognizes the polypyrimidine tract (Py-tract) consensus sequence of the pre-mRNA using two RNA recognition motifs (RRMs), the most prevalent class of eukaryotic RNA binding domain. The Py-tracts of higher eukaryotic pre-mRNAs are often interrupted with purines, yet U2AF65 must identify these degenerate Py-tracts for accurate pre-mRNA splicing. Previously, the structure of a U2AF65 variant in complex with polyuridine RNA suggested that rearrangement of flexible side chains or bound water molecules may contribute to degenerate Py-tract recognition by U2AF65. Here, the X-ray structure of the N-terminal RRM domain of U2AF65 (RRM1) is described at 1.47Å resolution in the absence of RNA. Notably, RNA-binding by U2AF65 selectively stabilizes pre-existing alternative conformations of three side chains located at the RNA interface (Arg150, Lys225, Arg227). Additionally, a flexible loop connecting the β2/β3 strands undergoes a conformational change to interact with the RNA. These pre-existing alternative conformations may contribute to the ability of U2AF65 to recognize a variety of Py-tract sequences. This rare, high resolution view of an important member of the RRM class of RNA binding domains highlights the role of alternative side chain conformations in RNA recognition.
Keywords: Protein-RNA recognition, RRM, alternative conformations, U2AF, pre-mRNA splicing
PDB reference: U2AF65, RNA recognition motif 1, 2HZC (updated coordinates), 2FZRsf
The RNA recognition motif (RRM) is one of the most abundant types of eukaryotic RNA binding domains, as exemplified by more than ~600 nonredundant human proteins documented by Pfam1. To date, more than 40 different structures of RRM-containing proteins have been determined, including at least ten distinct complexes with RNA. These structures establish an ~80 residue core composed of four β-strands packed against two α-helices. Two ribonucleoprotein consensus sequences on the central β-strands (β3 and β1, respectively called RNP1 and RNP2) display aromatic and basic residues that are important for RNA affinity. The β-sheet surface and additional N- or C-terminal extensions or loops of the RRM interact with the RNA strand. In many cases, the presence of several RRMs within a given polypeptide contributes additional sequence-specificity. Despite these established themes of RNA recognition by RRM-containing proteins, RRM/RNA specificity is far from predictable, and efforts are ongoing to establish the structural and energetic basis of this important type of RNA recognition.
Few high resolution (<1.5Å resolution) X-ray structures of RRM-containing proteins are available. Thus, little is known concerning the roles of alternative conformational states and hydration by bound water molecules during RRM/RNA recognition. In a singular example, the structure of a fragment of hnRNP A1 containing two RRMs was determined at 1.1Å resolution in the absence of nucleic acid2. Spatially-correlated alternative conformations for three residues in the RNP motifs of the N-terminal RRM of hnRNP A1 were observed in the absence of nucleic acid. When compared with the 2.1Å resolution structure bound to DNA ligand3, the presence of the nucleic acid sterically precludes one set of these alternative conformations. Without additional high resolution structural examples of RRMs, it is difficult to conclude whether selective ordering of pre-existing alternative conformations by RNA binding represents a recurring theme of nucleic acid recognition by RRM-containing proteins.
The importance of specific RNA interactions during RNA processing is illustrated by the substantial number of human genetic diseases associated with errors in pre-mRNA splicing4. During pre-mRNA splicing, the essential splicing factor U2AF65 recognizes the Py-tract consensus sequence near the 3′ splice site of the pre-mRNA. U2AF65 subsequently facilitates stable association of the pre-mRNA with the U2 small nuclear ribonucleoprotein particle (snRNP), a core component of the spliceosome5; 6. As for many proteins involved in RNA processing, two consecutive RRMs (RRM1 and RRM2) guide Py-tract recognition by U2AF657; 8. In addition to recognizing the Py-tract, the N-terminal U2AF65-RRM1 interacts with U2AF Associated Protein 56KD (UAP56), an RNP unwindase that is required for stable association of the U2 snRNP with the pre-mRNA9. Nuclear magnetic resonance (NMR) structures of the individual U2AF65 RRMs reveal the folds of these domains in the absence of nucleic acid10. Recently, the X-ray structure of a U2AF65 fragment in complex with polyuridine, and an accompanying analysis of site-directed mutant proteins, established the importance of RRM1 residues for polyuridine recognition11.
The ability of a Py-tract to direct splicing of a nearby pre-mRNA splice site is proportional to the number of consecutive uridines in the sequence12; 13. However, the Py-tracts of higher eukaryotic pre-mRNAs frequently are interrupted by cytosines, adenosines, or guanosines (50% Uri, 30% Cyt, 10% Ade, 10% Gua)14. Thus, U2AF65 is faced with the problem of specifying Py-tracts to ensure accurate splicing, yet tolerating a variety of natural Py-tract sequences15. The structure of the U2AF65 fragment/polyuridine complex suggests that flexible side chains and bound water molecules may rearrange during recognition of degenerate Py-tract sequences. Here, the X-ray structure of U2AF65-RRM1 at 1.47Å resolution provides a detailed view of the conformation and hydration in the absence of RNA, adding to a miniscule database of high resolution RRM structures. This structure suggests that flexible conformations of three side chains and a loop region of RRM1 may contribute to the ability of U2AF65 to tolerate Py-tracts interrupted by purines. In the one other available atomic resolution structure of an RRM domain (hnRNP A1)2, alternative side chain conformations also participate in nucleic acid binding. Thus, the high resolution view of the U2AF65 RRM supports the possibility of a general role for pre-existing alternative conformations in nucleic acid recognition by RRM-containing proteins.
High resolution structure of U2AF65-RRM1
The structure of human U2AF65-RRM1 (residues 148 to 229) was determined by multiwavelength anomalous dispersion (MAD) phasing (Supplementary Table 1). The final U2AF65-RRM1 coordinates consist of 87 residues, including five N-terminal residues from the protease cleavage site (Gly-Pro-Leu-Gly-Ser), 193 water molecules, two zinc ions and a partial molecule of PEG MME 550 (Figure 1(a) and (b)). The secondary structural arrangement of U2AF65-RRM1 (βαββαβ) corresponds to the canonical RRM fold1, as previously noted10. When compared with other RRM structures determined in the absence of RNA using X-ray crystallography including U1A, Sex-lethal, and hnRNP A116; 17; 18 (Figure 1(c)), the structures match relatively well in the core of the fold (1.3–1.5Å r.m.s.d. between 58–67 matching Cα atoms). However, unusual features of U2AF65-RRM1 emerge, namely an additional C-terminal turn of the first α-helix, an unusually long loop (12 residues) connecting this α-helix and second β-strand (α1/β2), and an unusually short loop (four residues) connecting the second and third β-strands (β2/β3). The α1/β2 loop is distant from the RNA binding site (20Å from the closest nucleotide, Uri7). In contrast, the minimal length of the β2/β3 loop is important for Py-tract recognition, as described below.
Figure 1.
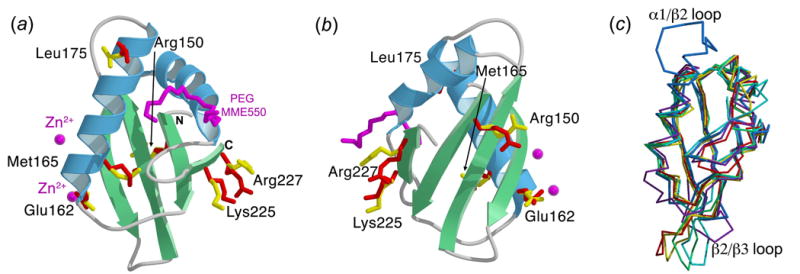
Overall structure of U2AF65-RRM1. Alternative conformations are shown as ball-and-stick representations with the major conformation in yellow and the minor conformation in red. (a) Ribbon diagram of U2AF65-RRM1 with α-helices colored in blue and β-strands in green. Bound PEG MME550 and zinc ions from the crystallization solution are shown in magenta. (b) View as in (a), rotated 180º about the y-axis. (c) Backbone trace representations of U2AF65-RRM1 (blue) compared with X-ray structures of other RRMs determined in the absence of RNA, including the N-terminal RRM of U1A24 (purple), N- and C-terminal RRMs of Sex-lethal16 (red and green, respectively), and N-and C-terminal RRMs of hnRNP A117 (yellow and cyan, respectively). The unique α1/β2 loop and β2/β3 loop of U2AF65-RRM1 are labeled. Images created using MolScript25, Bobscript26, and Raster3D27.
The relatively substantial differences between the X-ray and NMR10 structures of the apo-U2AF65-RRM1 (2.2Å r.m.s.d. for 82 matching Cα atoms) are comparable to differences between other structures determined by both X-ray and NMR methods19. The α1/β2 loop is the most divergent region between the X-ray and NMR structures of apo-U2AF65-RRM1, despite qualitatively similar hydrophobic interactions among Met173, Leu178, Thr179, and Pro185 side chains. Accordingly, the α1/β2 loop conformations are nearly identical between the apo- and RNA-bound X-ray structures, and are well defined (17Å2 B-factors for residues 179–184 compared with 22Å2 overall for apo-U2AF65-RRM1) (Figure 2(a)). The α1/β2 loop is located on the opposite face of the RRM compared with the RNA binding surface, where its distinctive, well-defined shape for docking of other splicing factors such as UAP569.
Figure 2.
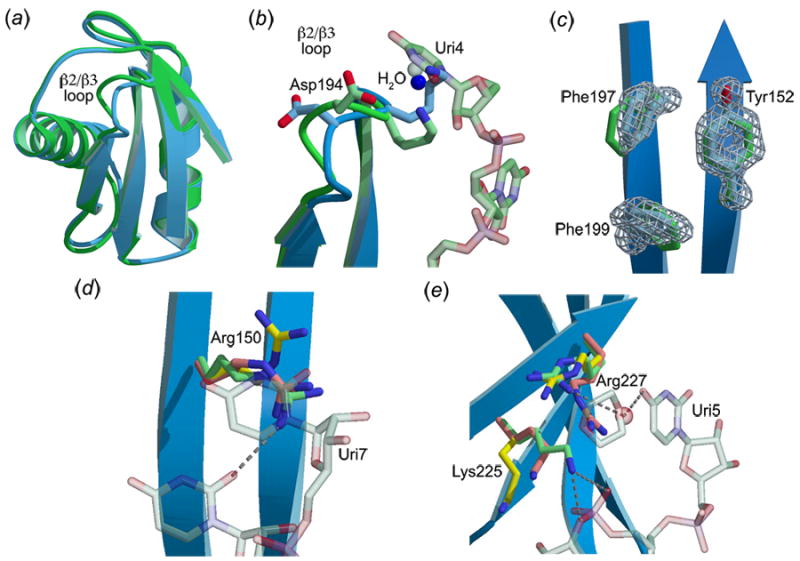
Comparison of apo- and RNA-bound U2AF65-RRM1. U2AF65-RRM1 in the presence (green) or absence (blue) of RNA ligand. When alternative conformations are present, major conformations of the apo-RRM1 side chains are colored yellow, minor conformations are colored orange. (a) Superimposed ribbon diagrams. (b) View of the α1/β2 loop. (c) Comparison of aromatic residues (Phe197, Phe199, Tyr152) in the RNP motifs. Sigma-A weighted, |Fo|-|Fc| omit electron density for side chain atoms was calculated using Shelxpro28 and is shown at 5σ contour level. (d) View of Arg150. (e) View of Lys225 and Arg227.
Comparison of the overall apo- and RNA-bound U2AF65-RRM1 structures
The major differences between the apo- and RNA-bound U2AF65 RRM1 structures are summarized in Table 1. Overall, the positions of the U2AF65-RRM1 backbone atoms remain largely unchanged by association with RNA (0.6Å r.m.s.d. for 82 matching Cα atoms) (Figure 2(a)). The β2/β3 loop is the most variable region between the apo- and RNA-bound U2AF65-RRM1 X-ray structures (Figure 2(b)). Despite its minimal length, the β2/β3 loop is highly flexible, as reflected by poorly defined electron density and the highest temperature factors of the apo-U2AF65-RRM1 structure (48Å2 average for residues 192 to 196 compared with 22Å2 overall). The RNA strand abuts the β2/β3 loop in the co-crystal structure of the U2AF65-RRM1 with polyuridine11, securing its conformation and explaining the unusually short length of this loop compared with other known RRM structures. When the apo-U2AF65-RRM1 structure is superimposed on the U2AF65-RRM1/polyuridine structure, Lys195, from the β2/β3 loop, overlaps a uridine (Uri4). The backbone of the β2/β3 loop avoids this potential steric clash by shifting 2Å in the presence of the RNA strand, enabling Lys195 to form both direct and water-mediated hydrogen bonds with Uri3 and Uri4. The conformational change of the loop also rearranges the backbone of Asp194 to provide a water-mediated hydrogen bond with Uri4 (Figure 2(b)).
Table 1.
Major differences between the apo- and RNA-bound U2AF65 RRM1 structures a
| Region | apo-U2AF65RRM1 | RNA-bound U2AF65RRM1 | Figure |
|---|---|---|---|
| β2/β3 loop | Poorly ordered | Well ordered, shifts to interact with RNA | Figure 1(b) |
| Water molecules near β2/β3 loop | Prepositioned for RNA interactions | Interacts with Uri3 & Uri4 | Figure 1(b); Supp. Figure 1 |
| Aromatic residues in RNP motifs | Well ordered | Slight shifts to stack with RNA bases | Figure 2(c) |
| Arg150 | Major conformation interacts with Gly146 & Glu201; Minor conformation stacks with symmetry-related Phe158 and is most frequent rotamer (44%) | Only minor conformation of apo-RRM1-Arg150 is observed, which stacks with RNA base | Figure 2(d) |
| Lys225 | Major conformation is most frequent rotamer (42%); Minor conformation is second most frequent rotamer (25%) | Only minor conformation of apo-RRM1-Lys225 is observed, which interacts with RNA | Figure 2(e) |
| Arg227 | Major conformation interacts with Glu215; Minor conformation is the most frequent rotamer (44%) | Only major conformation of apo-RRM1-Arg227 is observed, which interacts indirectly with RNA via water molecule, and directly with Glu215 & dioxane from cryosolvent; Minor conformation would be too close to RNA-bound Lys225 conformation (3.3Å Nζ-NH1 distance) | Figure 2(e) |
Descriptions of minor alternative conformations are distinguished by italics.
Although the backbones of the apo- compared with the RNA-bound U2AF65-RRM1 structures are nearly identical in the β-sheet region that serves as the major interface with the RNA strand, conformational differences are observed for several of the side chains. Two phenylalanines in the RNP1 motif show slight (<1Å) torsional shifts that accommodate stacking with either the RNA backbone (Uri5/Phe197) or the base (Uri6/Phe199) (Figure 2(c)). Correlated alternative side chain conformations are observed for two phenylalanine residues in the RNP motifs of the apo-hnRNP A1 structure. However, alternative conformations of the RNP motifs may not be a general characteristic of RRMs, as the corresponding U2AF65-RRM1 residues (Tyr152 and Phe199) display discrete, well-defined electron density (Figure 2(c)). Apart from the β2/β3 loop, the major differences in the RNA binding surface between the apo- and RNA-bound U2AF65-RRM1 structures are observed among side chains that display alternative conformations in the apo-U2AF65-RRM1 (Arg150, Lys225, and Arg227). Although the resolution of the RNA-bound structure is lower (2.5Å) than the present apo-structure (1.47Å), the presence of RNA-induced contacts or steric clashes clearly rule out one of the alternative side chain conformations that pre-exist in the absence of RNA (Figure 2(d–e)). These alternative conformations are described in detail below.
Comparison of bound water molecules in the apo- and RNA-bound U2AF65-RRM1 structures
The lower resolution of the RNA-bound U2AF65-RRM1 hinders a comprehensive analysis of the positions of water molecules bound to the protein or RNA surface. However, five water molecules interacting with four of the seven bound nucleotides (Uri3, Uri4, Uri5, and Uri7) were observed in the electron density for the U2AF65/polyuridine complex11. One of these water molecules is pre-positioned in the apo-U2AF65-RRM1 structure for water-mediated interactions with the RNA strand (Figure 2(b), Supplementary Figure 1 (a, b)). This water is held securely in place by the backbone carboxamides of Asn155, Asn186, and Lys195 of the apo-U2AF65-RRM1 (B-factor 31Å2). In the presence of polyuridine RNA, the hydrogen bond with the Lys195 carbonyl is replaced by hydrogen bonds with the 2′ hydroxyl group of Uri3 and a phosphate oxygen of Uri4, and with the side chain oxygen of the rearranged Asn196 conformation in the β2/β3 loop. A second water molecule mediates interactions between the edge of the Uri3 base and residues in the C-terminal U2AF65-RRM (RRM2) (Supplementary Figure 1 (c, d)). In the absence of U2AF65-RRM2 and Uri3, a nearby water molecule (1.7Å distance between water molecules in the absence and presence of RNA) interacts with residues in RRM1 (Asn155, Gln222, and the carbonyl of Ile156). In the intact RNA binding domain containing both RRM1 and RRM2, this water molecule may be pre-positioned to interact with Uri3.
The three remaining water molecules that interact with the polyuridine RNA are absent in the apo-RRM1 structure. In the presence of RNA, a water molecule interacts with the Uri4 base and is held in place by the RNA-bound conformation of the β2/β3 loop, in particular the carbonyl of Asp194 and side chain of Lys195. In the absence of RNA, the altered conformation of the loop and the Asp194 side chain releases the bound water (Figure 2(b)). The water molecule that interacts with Uri5 is replaced by the alternative conformation of Arg227 in the apo-U2AF65-RRM1 structure (Figure 2(e)), although the possibility cannot be ruled out that this water represents a minor alternative conformation of Arg227 at the lower resolution of the U2AF65/polyuridine structure. The third water molecule binds the edge of the Uri7 base, but lacks direct contacts with U2AF65 and is not observed in the absence of the uridine.
Overview of alternative side chain conformations
The high resolution of the apo-U2AF65-RRM1 X-ray structure revealed that six side chains (Arg150, Glu162, Met165, Leu175, Lys225, Arg227), or 7% of the total residues in the domain, have two alternative conformations with nearly equivalent occupancies (respectively 0.55, 0.66, 0.50, 0.57, 0.68, 0.62, for the major conformations) (Figure 3). Only one of these (Met165) is found in the protein interior. The alternative conformations of Met165 seek to fill a hydrophobic pocket in the protein core (Figure 3(c)). Accordingly, Met165 of human U2AF65 is replaced with slightly bulkier valine or phenylalanine residues in Arabidopsis or fission yeast U2AF65, respectively, which may improve the hydrophobic packing. Remaining alternative conformations are located on the surface of the protein and are exposed to solvent in the absence of bound RNA. Among these, Glu162 is influenced by the crystal packing environment, namely by coordination with a zinc ion from the crystallization solution. The alternative conformations of Leu175 may also be influenced by the crystal packing environment, since the major conformation is closest to Asp206 of a symmetry-related molecule (5.5Å Leu175a-Cδ2---Asp206′-Cβ), whereas the other is closer to a carbon atom of a symmetry-related PEG molecule (5.5Å Leu175b-Cδ1---PEG′-C6). In contrast, Arg150, Lys225, and Arg227 are located at or near the RNA interface. Comparison of the apo-U2AF65-RRM1 with the RNA-bound structure establishes that RNA selectively stabilizes a subset of these side chain conformations, as described below and summarized in Table 1.
Figure 3.
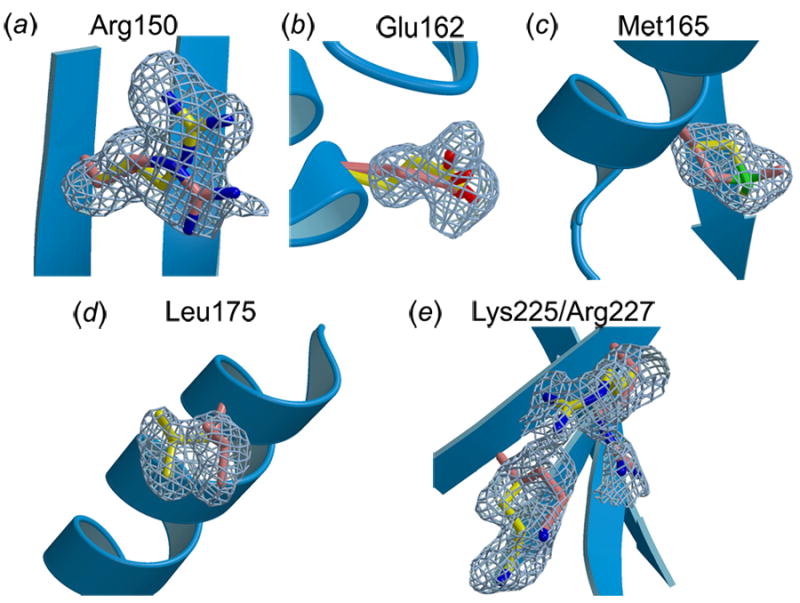
Alternative conformations of U2AF65-RRM1 side chains with sigma-A weighted, |Fo|-|Fc| omit electron density shown at 2σ contour level, colored as in Figure 2. (a) Arg150, (b) Glu162, (c) Met165, (d) Leu175, (e) Lys225 and Arg227.
Alternative side chain conformations at the RNA binding surface
The most striking of the RNA-dependent conformations involves Arg150 at the 3′ terminus of the RNA (Figure 2(d) and Figure 3(a)). In the apo-structure, one alternative conformation of Arg150 is located with the positively charged guanidinium group stacking against the negatively charged Glu201 side chain, and the NH1 atom donating hydrogen bonds to both a bound water molecule and the backbone carbonyl of Gly146. The second, slightly lower occupancy conformation is turned towards the body of the protein, and participates in water-mediated hydrogen bonds with the Gln190 side chain. The two alternative conformations have equivalent occupancies (0.55 and 0.45, respectively), and the minor conformation is the most frequently observed arginine rotamer in a database of protein structures (44% frequency)20. Arg150 fully adopts this latter conformation in the presence of the polyuridine RNA; in this position the guanidinium group is selectively stabilized by parallel aromatic stacking interactions with the Uri7 base, and a hydrogen bond with the O2 atom of Uri6.
Lys225 and Arg227 are located in the C-terminal β-strand of U2AF65-RRM1, near the 5′ end of the bound RNA strand (Figure 2(e) and Figure 3(e)). In the absence of RNA, the major Lys225 conformation (occupancy 0.7) is the most frequently observed lysine rotamer (42% frequency), and the minor Lys225 conformation (occupancy 0.3) is the second most frequently observed lysine rotamer (25% frequency)20. Association with RNA selectively stabilized the minor Lys225 alternative conformation, which also is extended to interact with the O3’ atom of Uri3 and the pro-S phosphate oxygen of Uri4. In the absence of RNA, the major Arg227 conformation (occupancy 0.6) is stabilized by interactions with Glu215 despite adopting an infrequently observed arginine rotamer (11% frequency), whereas the minor Arg227 conformation (occupancy 0.4) is the most frequently observed arginine rotamer (44% frequency)20. In the presence of RNA, movement of Lys225 forces Arg227 to fully adopt the major alternative conformation observed in the apo-structure, thereby avoiding uncomfortably close interactions with the similarly-charged Lys225 side chain (3.3 Å predicted Nζ-NH1 distance between RNA-bound Lys225 and the minor conformation of Arg227, compared with 5.0 Å distance in the absence of RNA) (Figure 2(e)). The RNA-bound Arg227 conformation participates in a water-mediated hydrogen bond with the Uri5 base. A nearby dioxane molecule from the crystallization solution of the RNA complex also influences the Arg227 conformation. With minor readjustment, the unselected Arg227 conformation in the apo-structure could participate directly in hydrogen bonds with the base of Uri5 without disturbing the Lys225/phosphate contacts, suggesting that this conformation may participate in RNA interactions in the absence of crystallization solution.
Implications for pre-mRNA splice site recognition
To fulfill its role as an essential splicing factor, U2AF65 must accurately specify the Py-tract and recruit the splicing machinery to the appropriate 3′ splice site. The majority of U2AF65-RRM1 residues adopt similar conformations in the presence and absence of the polyuridine ligand, providing a prearranged molecular shape that may contribute to the preference of U2AF65 for binding Py-tracts21; 22. However, U2AF65 also must adapt to deviations from the uridine-rich consensus sequence in most Py-tracts of higher eukaryotes14. The flexibility of side chains at the RNA interface is evident from three alternative conformations observed in this region of the apo-U2AF65-RRM1. Comparison with the co-crystal structure of the U2AF65 RRMs in complex with a polyuridine oligonucleotide reveals that the presence of bound uridines selectively stabilized a subset of these alternative conformations. In addition, the β2/β3 loop must rearrange to fit the path of the RNA strand. Importantly, the inherent flexibility of these side chains and β2/β3 loop suggest a possible mechanism for U2AF65 to tolerate degenerate Py-tracts by favoring different subsets of energetically-equivalent alternative conformations. For example, a larger purine base substituted for Uri7 would stack favorably with the alternative conformation of Arg150 that was eliminated by binding the polyuridine sequence. Furthermore, if cytosine or adenosine is substituted for Uri4, the Asn196 conformation observed in the β2/β3 loop of the apo-structure is pre-positioned to donate a favorable hydrogen bond to the exocyclic amine of the mutated base. Although these flexible U2AF65 RRM1 regions interact with defined nucleotides of the RNA strand, in practice it is difficult to predict which Py tract positions exhibit more tolerance for nucleotide substitutions, since U2AF65 binds natural Py tracts in multiple registers23.
Both available high resolution structures of RRM domains determined in the presence and absence of RNA (hnRNP A12 and now U2AF65 RRM1) reveal selective ordering of pre-existing alternative side chain conformations by RNA binding. Thus, the possible role of alternative conformations should be considered when predicting or interpreting the characteristics of RNA recognition by RRM-containing proteins, which has broad significance for the numerous human genetic diseases resulting from errors in RNA recognition by RRM-containing proteins4.
Acknowledgments
We thank S.K. Burley, in whose laboratory this research was initiated, for support and advice. We thank the BioCARS staff for use of Station 14-ID-B at the Advanced Photon Source, which receives support (through grant RR07707) from the National Center for Research Resources of the National Institutes of Health. We thank C. Wolberger, L.M. Amzel, and D. Leahy for advice and generous access to X-ray equipment. M.M. Benning (Bruker, AXS) and J.D. Ferrara (MSC) assisted with collection of high resolution native data sets, and J. Bair assisted with protein production. K.R.T. was supported in part by a Training Grant (T32 GM08403) from the National Institutes of Health (NIH). The laboratory of C.L.K. is supported by the NIH (GM070503).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–31. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 2.Vitali J, Ding J, Jiang J, Zhang Y, Krainer AR, Xu RM. Correlated alternative side chain conformations in the RNA recognition motif of heterogeneous nuclear ribonucleoprotein A1. Nucleic Acids Res. 2002;30:1531–8. doi: 10.1093/nar/30.7.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding J, Hayashi MK, Zhang Y, Manche L, Krainer AR, Xu RM. Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev. 1999;13:1102–15. doi: 10.1101/gad.13.9.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–37. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 5.Ruskin B, Zamore PD, Green MR. A factor, U2AF, is required for U2 snRNP binding and splicing complex assembly. Cell. 1988;52:207–219. doi: 10.1016/0092-8674(88)90509-0. [DOI] [PubMed] [Google Scholar]
- 6.Zamore PD, Green MR. Biochemical characterization of U2 snRNP auxiliary factor: an essential pre-mRNA splicing factor with a novel intranuclear distribution. EMBO J. 1991;10:207–14. doi: 10.1002/j.1460-2075.1991.tb07937.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zamore PD, Patton JG, Green MR. Cloning and domain structure of the mammalian splicing factor U2AF. Nature. 1992;355:609–14. doi: 10.1038/355609a0. [DOI] [PubMed] [Google Scholar]
- 8.Banerjee H, Rahn A, Gawande B, Guth S, Valcarcel J, Singh R. The conserved RNA recognition motif 3 of U2 snRNA auxiliary factor (U2AF65) is essential in vivo but dispensable for activity in vitro. RNA. 2004;10:240–253. doi: 10.1261/rna.5153204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleckner J, Zhang M, Valcarcel J, Green MR. U2AF65 recruits a novel human DEAD box protein required for the U2 snRNP-branchpoint interaction. Genes Dev. 1997;11:1864–72. doi: 10.1101/gad.11.14.1864. [DOI] [PubMed] [Google Scholar]
- 10.Ito T, Muto Y, Green MR, Yokoyama S. Solution structures of the first and second RNA-binding domains of human U2 small nuclear ribonucleoprotein particle auxiliary factor (U2AF65) EMBO J. 1999;18:4523–4534. doi: 10.1093/emboj/18.16.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sickmier EA, Frato KE, Shen H, Paranawithana SR, Green MR, Kielkopf CL. Structural basis of polypyrimidine tract recognition by the essential pre-mRNA splicing factor, U2AF65. Mol Cell. 2006;23:49–59. doi: 10.1016/j.molcel.2006.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coolidge CJ, Seely RJ, Patton JG. Functional analysis of the polypyrimidine tract in pre-mRNA splicing. Nucleic Acids Res. 1997;25:888–96. doi: 10.1093/nar/25.4.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reed R. The organization of 3′ splice-site sequences in mammalian introns. Genes Dev. 1989;3:2113–2123. doi: 10.1101/gad.3.12b.2113. [DOI] [PubMed] [Google Scholar]
- 14.Senapathy P, Shapiro MB, Harris NL. Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol. 1990;183:252–78. doi: 10.1016/0076-6879(90)83018-5. [DOI] [PubMed] [Google Scholar]
- 15.Zhang M, Zamore PD, Carmo-Fonseca M, Lamond AI, Green MR. Cloning and intracellular localization of the U2 small nuclear ribonucleoprotein auxiliary factor small subunit. Proc Natl Acad Sci U S A. 1992;89:8769–8773. doi: 10.1073/pnas.89.18.8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crowder SM, Kanaar R, Rio DC, Alber T. Absence of interdomain contacts in the crystal structure of the RNA recognition motifs of Sex-lethal. Proc Natl Acad Sci U S A. 1999;96:4892–7. doi: 10.1073/pnas.96.9.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shamoo Y, Krueger U, Rice LM, Williams KR, Steitz TA. Crystal structure of the two RNA binding domains of human hnRNP A1 at 1.75 A resolution. Nat Struct Biol. 1997;4:215–22. doi: 10.1038/nsb0397-215. [DOI] [PubMed] [Google Scholar]
- 18.Nagai K, Oubridge C, Jessen TH, Li J, Evans PR. Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature. 1990;348:515–520. doi: 10.1038/348515a0. [DOI] [PubMed] [Google Scholar]
- 19.Garbuzynskiy SO, Melnik BS, Lobanov MY, Finkelstein AV, Galzitskaya OV. Comparison of X-ray and NMR structures: Is there a systematic difference in residue contacts between X-ray- and NMR-resolved protein structures? Proteins: Structure, Function, and Bioinformatics. 2005;60:139–147. doi: 10.1002/prot.20491. [DOI] [PubMed] [Google Scholar]
- 20.Kleywegt GJ, Jones TA. Databases in protein crystallography. Acta Crystallogr D Biol Crystallogr. 1998;54:1119–31. doi: 10.1107/s0907444998007100. [DOI] [PubMed] [Google Scholar]
- 21.Singh R, Valcarcel J, Green MR. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 22.Singh R, Banerjee H, Green MR. Differential recognition of the polypyrimidine-tract by the general splicing factor U2AF65 and the splicing repressor Sex-lethal. RNA. 2000;6:901–11. doi: 10.1017/s1355838200000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banerjee H, Rahn A, Davis W, Singh R. Sex lethal and U2 small nuclear ribonucleoprotein auxiliary factor (U2AF65) recognize polypyrimidine tracts using multiple modes of binding. RNA. 2003;9:88–99. doi: 10.1261/rna.2131603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagai K, Oubridge C, Jessen TH, Li J, Evans PR. Crystal structure of the RNA-binding domain of the U1 small nuclear ribonucleoprotein A. Nature. 1990;348:515–20. doi: 10.1038/348515a0. [DOI] [PubMed] [Google Scholar]
- 25.Kuralis PJ. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 26.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. 1999;D55:938–40. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 27.Merritt EA, Bacon DJ. Raster3D - photorealistic molecular graphics. Methods Enzym. 1997:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 28.Sheldrick GM, Schneider TR. SHELX-97: high resolution refinement. Methods Enzymol. 1997;276:319–343. [PubMed] [Google Scholar]