

Abstract
IL-23 induces IL-17 production in activated CD4+ T cells and participates in host defense against many encapsulated bacteria. However, whether IL-23/IL-17 axis contributes to a Mycoplasma pneumoniae (Mp)-induced lung inflammation (e.g., neutrophils) has not been addressed. Using an acute respiratory Mp infection murine model, we found significantly up-regulated lung IL-23p19 mRNA in the early phase of infection (4 h), and alveolar macrophages were an important cell source of Mp-induced IL-23. We further showed that Mp significantly increased IL-17 protein levels in bronchoalveolar lavage (BAL). Lung gene expression of IL-17, IL-17C and IL-17F was also markedly up-regulated by Mp in vivo. IL-17 and IL-17F were found to be derived mainly from lung CD4+ T cells, and were increased upon IL-23 stimulation in vitro. In vivo blocking of IL-23p19 alone or in combination with IL-23/IL-12p40 resulted in a significant reduction of Mp-induced IL-17 protein and IL-17/IL-17F mRNA expression, which was accompanied by a trend toward reduced lung neutrophil recruitment, BAL neutrophil activity, and Mp clearance. However, IL-23 neutralization had no effect on Mp-induced lung IL-17C mRNA expression. These results demonstrate that IL-17/IL-17F production is IL-23-dependent in an acute Mp infection, and contributes to neutrophil recruitment and activity in lung defense against the infection.
Keywords: M. pneumoniae, IL-23, IL-17, IL-17F, neutrophil
1. Introduction
Mycoplasmas represent the smallest (50–300 nm in diameter) organisms capable of self-replication in cell-free media. These organisms are unique in that they lack a cell wall, a feature largely responsible for their biologic properties such as lack of a reaction to Gram staining, and insensitivity to many commonly prescribed antimicrobial agents [1]. Mycoplasma pneumoniae (Mp), one of the human mycoplasma species, resides extracellularly in the respiratory tract mucosa, and represents one of the most common causes of community-acquired pneumonia [2]. Several studies have shown that Mp infection induces the release of proinflammatory cytokines relevant to exacerbation of chronic pulmonary diseases such as asthma and COPD [3–4]. Previous studies have demonstrated that respiratory Mp infection is characterized by lung neutrophilic inflammation along with lung lymphocyte (e.g., CD4+ T cells) infiltrate [5–8]. However, the molecular mechanisms by which Mp induces CD4 T cell and neutrophil influx into lung tissues remain poorly understood.
IL-17 family is a recently described group of cytokines including IL-17 (or IL-17A), and IL-17B–F. IL-17 and IL-17F are mainly expressed by activated CD4+ T cells and involved in host innate immune response such as neutrophil recruitment and activity [9]. Recent publications suggest that IL-17 production from activated T cells is inducible upon IL-23 stimulation [10–12]. IL-23 is a novel member of the IL-12 cytokine family and is composed of a unique p19 subunit and an identical p40 subunit to IL-12. Activated macrophages and dendritic cells secrete IL-23 in response to environmental danger signals [13]. It should be emphasized that although IL-23 belongs to the IL-12 family, IL-23 has been shown to exert the opposite function in regulating IL-17 as compared to IL-12. Specifically, while IL-23 promotes IL-17 production, IL-12 was shown to inhibit IL-17 elaboration in both human and mouse systems [10, 14, 15].
Previous studies have shown that IL-23/IL-17 axis may be important in host defense against Gram-positive and Gram-negative bacterial infections (e.g. K. pneumoniae, T. gondii and C. neoformans) [11, 16, 17]. However, whether IL-23/IL-17 axis contributes to an Mp-induced lung inflammation (e.g., neutrophils) has not been addressed. We therefore determined whether respiratory Mp infection in mice activates innate immune cells (i.e., alveolar macrophages) to produce IL-23, thus increasing production of IL-17 and its family members by pulmonary CD4+ T cells. We then neutralized IL-23p19 or IL-23p19 plus IL-23/IL-12p40 to further investigate the role of IL-23 in modulating IL-17 family members and lung neutrophil function.
2. Materials and Methods
2.1. Mice
Animal experiments were approved by our Institutional Animal Care and Use Committee. Female BALB/c mice (8–12 wk old) were obtained from the Jackson Laboratory (Bar Harbor, ME, US). All the mice were quarantined for 4 weeks before the experiment and bled to establish that they were virus and Mycoplasma pulmonis free.
2.2. Mycoplasma pneumoniae (Mp) inoculation
Mp (FH strain, ATCC 15531) was prepared in saline to approximately yield 1 × 107 colony forming units (CFUs)/50 μl. On day 0, mice were anesthetized i.p. with 0.25 g/kg body weight of Avertine (2, 2, 2- Tribromoethanol dissolved in liquid tert-Amyl alcohol at 0.025 g/ml, Sigma, St. Louis, MO, US). In the infected groups (n=10/group), mice were inoculated intranasally with 50 μl of Mp at 1 × 107 CFUs/mouse. A 50-μl of saline was similarly given to the control mice (n=5/group). Mice were sacrificed at 4 h, days 1, 3 and 7.
2.3. Bronchoalveolar lavage (BAL) collection
Mice were euthanized by exposure to CO2. The lung was lavaged with 1ml saline. 10 μl of BAL was used for Mp culture. Cell-free BAL fluid was stored at –80°C for cytokine or myeloperioxidase (MPO) activity assay. Cytospins of BAL cells were stained with Diff-Quick Kit (IMEB INC., San Marcos, CA, US), and cell differentials were determined as percentage of 500 counted cells.
2.4. Primary alveolar macrophages (pAMs) enrichment and in vitro Mp infection
We determined the IL-23p19 mRNA levels in freshly isolated pAMs obtained from Mp- or saline-treated mice (n=5/group) at 4 h and 24 h post infection. BAL cells were pooled in the same group and resuspended in RPMI-1640 medium with 10% FBS, and seeded in a 24-well culture plate (2 × 104 cells/well) to incubate for 2 h at 37°C, 5% CO2. The adherent cells were harvested in TRIzol (Invitrogen, Carlsbad, CA, US) for RNA extraction.
To confirm the direct effects of Mp on IL-23p19 mRNA by alveolar macrophages, pAMS were isolated from pooled BAL cells of saline-treated mice at 24 h. The adherent cells were treated in the absence or presence of Mp (10 CFUs/cell) for 4 h and 24 h, and were then collected in TRIzol for RNA extraction.
In all the aforementioned experiments, more than 95% of adherent cells were identified as macrophages under the light microscope on 8-well chamber slides with Diff-Quick staining (IMEB INC., San Marcos, CA, US).
2.5. Lung tissue allocation
After BAL, lungs were perfused with PBS and excised. The left lung was processed for H&E staining, Mp culture and RNA extraction. The whole right lung tissues were for CD4+ T cell isolation.
2.6. Pulmonary CD4+ T cell isolation
Lung digestions were performed as described [18]. Briefly, minced lungs were exposed to a 20 ml enzymatic mixture containing 6 units of Dispase II (Roche Diagnostics, Indianapolis, Indiana, US), 2 μg of Collagenase II and 2 μg of Collagenase IV (Sigma, St. Louis, MO, US) at 37°C for 75 min. Lung tissues from the same group of mice were pooled and mashed through a Cellector™ tissue sieve (Bellco Glass Inc. Vineland, NJ, US) to obtain single-cell suspensions. CD4+ T cells were purified using a CD4 T cell isolation kit and Mini-MACS columns (Miltenyi Biotec., Auburn, CA, US). FACS confirmed the purity of CD4+ T cells to be ≤95%.
2.7. In vitro recombinant mouse IL-23 (rmIL-23) stimulation of pulmonary CD4+ T cells
To determine IL-23 dose response and time course on IL-17 expression, pulmonary CD4+ T cells from saline-treated mice post 24 h were seeded at 2 × 105 cells/well into a 96-well culture plate and incubated with rmIL-23 (0.1 – 1000 ng/ml, Sf21-derived, R&D systems, Minneapolis, MN, US) for 24 h and 48 h. Cell supernatants were harvested for IL-17 ELISA.
To examine whether CD4+ T cells from naïve (saline treatment, n=5/group) or Mp-infected mice (n=10/group) differed in response to rmIL-23, CD4+ T cells isolated from saline- and Mp-treated mice at all the given time points were incubated in the absence or presence of 10 ng/ml rmIL-23 for 24 h and 48 h. Cell supernatants were harvested for IL-17 ELISA and cells were lysed in TRIzol. To further reproduce and confirm the effects of rmIL-23 on IL-17 expression, three independent experiments were repeated using CD4+ T cells from saline- and Mp-treated mice at 24 h.
2.8. IL-23 neutralization in mice
Sf21-derived goat anti-mouse IL-23p19 polyclonal antibody, rat anti-mouse IL-23/IL-12 p40 monoclonal antibody, normal goat IgG (purified from naïve goat serum, control for goat anti-mouse IL-23p19 antibody) and normal rat IgG2A antibody (produced from a hybridoma resulting from the fusion of a mouse myeloma with B cells obtained from a rat immunized with purified KLH, control for rat anti-mouse IL-23/IL-12p40 antibody) were purchased from R&D Systems (Minneapolis, MN, US). R&D system has determined the neutralizing activity of the anti-IL-23 p19 Ab in vitro by measuring IL-23-dependent production of IL-17 in mouse splenocytes. All the antibodies (Abs) were diluted in PBS for following treatments: group 1, anti-mouse IL-23p19 Ab + Mp; group 2, normal goat IgG (control for group 1) + Mp; group 3, (anti-mouse IL-23p19 Ab + anti-mouse IL-23/IL-12 p40 Ab) + Mp; and group 4, normal goat IgG + normal rat IgG2A (control for group 3) + Mp. Mice (n=5–8/group) were treated intranasally with Abs 2 h prior to Mp (1 × 107 CFU per mouse), and sacrificed after 24 h to collect lung and BAL samples for detecting IL-17 family members, Mp clearance and neutrophil activity.
2.9. ELISA
IL-17 was detected using the Quantikine® Mouse IL-17 Immunoassay (R&D Systems, Minneapolis, MN, US). The limit of detection was 5 pg/ml.
2.10. Real-time RT-PCR
Total RNA was extracted and treated with DNase I (Ambion, Austin, TX, US). Reverse transcription was performed using 1 μg of total RNA and random hexamers in a 50-μl reaction according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA, US). The mouse IL-23p19, IL-17, IL-17C and IL-17F primers and probes were designed using the Primer Express software (Applied Biosystems) and listed in Table 1.
Table 1.
Quantitative real-time PCR Primers and probe sequences
| Cytokines | Genbank Accession# | Sequences |
|---|---|---|
| IL-23p19 | NM_031252 | Forward primer: 5′-TGTGCCCCGTATCCAGTGT-3′
Reverse primer: 5′-CGGATCCTTTGCAAGCAGAA-3′ Probe: 5′-TGTGACCCACAAGGACTCAAGGACAACA-3′ |
| IL-17 | U43088 | Forward primer: 5′-ACCGCAATGAAGACCCTGAT-3′
Reverse primer: 5′-TCCCTCCGCATTGACACA-3′ Probe: 5′-CTGGGAAGCTCAGTGCCGCCAC-3′ |
| IL-17C | AF458061 | Forward primer: 5′-CCATGGAGATATCGCATCGA-3′
Reverse primer: 5′-GCATCCACGACACAAGCATT-3′ Probe: 5′-AGAACCGCTACCCACAGAAGCTGGC-3′ |
| IL-17F | AF458064 | Forward primer: 5′-CTGAGGCCCAGTGCAGACA-3′
Reverse primer: 5′-GCTGAATGGCGACGGAGTT-3′ Probe: 5′-CCCAGGGTCAGGAAGACAGCACCAT-3′ |
Real-time PCR was performed on the ABI Prism® 7700 sequence detection system (Applied Biosystems). The 25-μl PCR contained 30 ng of cDNA, 100 nM fluogenic probe, and 100 nM primers and other components from the TaqMan® RT-PCR kit (Applied Biosystems). 18S rRNA was evaluated as internal positive control. The comparative Ct method was used to demonstrate the relative mRNA levels of target genes.
2.11. Myeloperoxidase (MPO) activity assay
MPO is mainly released by neutrophils and has been used as an index of tissue neutrophil levels [19]. Thus, MPO activity levels in lung homogenates and BAL fluid were measured to reflect neutrophil accumulation and activity, respectively. MPO activity in lungs and BAL fluid were measured as described [20]. Lung tissues were homogenized in 50 mM potassium phosphate buffer (50 mg of tissue/ml) containing 0.5% hexadecyltrimethylammonium bromide (HTAB, Sigma). Supernatants were collected after centrifugation at 20,000 g for 5 min. The assay solution was prepared freshly by mixing a 50 mM potassium phosphate buffer (pH 6.0) containing 0.167 mg/ml o-dianisidine dihydrochloride and 0.0005% hydrogen peroxide (Sigma). Lung homogenates (7 μl) or cell-free BAL fluid (100 μl) were added to a 96-well plate. Then, 200 μl of the assay solution was added and mixed rapidly. The enzymatic activity was determined spectrophotometrically by measuring the change in absorbance at 450 nm using a plate reader. MPO activity was expressed in units per milligram of lung tissue or units per 106 neutrophils in BAL fluid.
2.12. Mp culture
10 μl of BAL or minced lung tissue (total size of 3 mm × 3 mm × 3 mm) was placed onto PPLO plates (Remel, Lenexa, KS, US) and incubated at 37°C, 5% CO2 for 7 days when CFUs were counted.
2.13. Statistical analysis
Normally distributed data are presented as means ± SEM and compared using the matched-pairs t test. Non-normally distributed data are expressed as medians with interquartile (25–75%) ranges and compared using the Wilcoxon rank-sum test. A value of p < 0.05 was considered significant.
3. Results
3.1. Inflammation profiles in BAL and lung tissues
As shown in Table 2, Mp infection markedly increased total leukocyte counts including neutrophils and lymphocytes in BAL, especially on days 1 and 3. The total macrophage counts were similar in infected and control groups. There was only < 0.2 % of eosinophils in any mouse BAL samples.
Table 2.
Bronchoalveolar lavage (BAL) cell profiles (×104 cells/ml) a
| Groups | White Blood cells | Neutrophils | Macrophages | Lymphocytes |
|---|---|---|---|---|
| 4 h | ||||
| Mp infection | 12.0 (6.0–18.5) | 10.2 (5.0–17.1) | 1.2 (0.9–1.6) | 0.2 (0.1–0.2) |
| Saline control | 1.5 (1.3–4.0) | 0.2 (0.1–0.2) | 1.3 (1.0–3.7) | 0.1 (0.1–0.1) |
| Day 1 | ||||
| Mp infection | 45.0 (38.3–74.5)* | 43.2 (36.8–70.4)* | 2.1 (0.9–3.9) | 0.3 (0.2–0.6)* |
| Saline control | 3.5 (1.0–5.8) | 0.1 (0.1–0.4) | 3.4 (0.9–5.3) | 0.02 (0.01–0.1) |
| Day 3 | ||||
| Mp infection | 29.8 (16.6–86.5)* | 20.6 (9.1–68.3)* | 8.3 (5.4–15.3) | 2.4 (1.9–3.6)* |
| Saline control | 8.0 (6.5–10.3) | 0.1 (0–0.1) | 7.8 (6.4–10.1) | 0.06 (0.03–0.2) |
| Day 7 | ||||
| Mp infection | 3.5 (1.9–6.0) | 0.1 (0.1–0.7) | 3.0 (1.6–4.8) | 0.2 (0.1–0.4) |
| Saline control | 2.5 (1.5–7.8) | 0.1 (0–0.1) | 2.4 (1.4–7.6) | 0.03 (0.01–0.1) |
M. pneumoniae (Mp) infection groups (n=10/group) and Saline control groups (n=5/group) at 4 h, days 1, 3 and 7. Data are expressed as medians (25–75% range).
p < 0.01, Wilcoxon rank-sum test between Mp group and Saline group at the same time point.
H&E stained lung tissues showed a similar inflammatory profile to BAL. The most intense inflammatory response such as large numbers of neutrophils and mononuclear cells was on days 1 and 3, but not day 7.
3.2. Mp increases IL-23 expression by alveolar macrophages
In Mp-infected mice, IL-23p19 mRNA levels in lung tissues were markedly increased (up to 100-fold) within 4 h post infection, remained elevated at 24 h and then decreased on days 3 and 7 compared to saline groups (Fig.1).
Fig. 1.
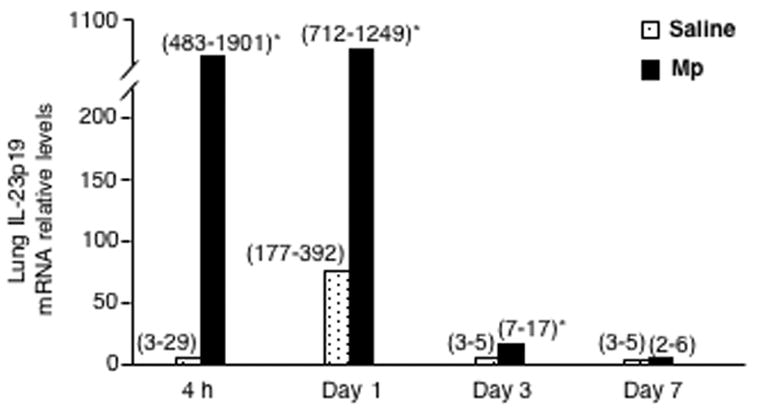
Lung tissue IL-23p19 mRNA levels. Lungs were obtained from saline-treated (n=5/group) and M. pneumoniae (Mp)-infected mice (n=10/group) at 4 h, days 1, 3 and 7 post saline or Mp. Data are expressed as medians (25–75% range). * p < 0.01, Wilcoxon rank-sum test between saline group and Mp group at the same time point.
As activated macrophages are one of the important cell sources of IL-23 in response to environmental danger signals [13], we isolated pAMs from BAL cells to determine if IL-23 p19 mRNA was increased by macrophages in response to Mp in vivo. It was found that freshly isolated pAMs from Mp-infected mice had 130 and 30 times higher levels of IL-23p19 mRNA than saline-treated mice at 4 h and 24 h post infection, respectively. Then, we confirmed the direct effects of Mp on IL-23 expression by infecting naïve pAMs (from saline-treated mice at 24 h) in vitro. With Mp infection, IL-23p19 mRNA levels were increased up to 770-fold and 17-fold compared to those of cell culture medium-treated (control) cells at 4 h and 24 h, respectively. Since a reliable IL-23 ELISA kit is not available, IL-23 protein was not measured.
3.3. Mp induces expression of IL-17 family members
The gene expressions of IL-17, IL-17C and IL-17F in lung tissues were markedly up-regulated by Mp in vivo as early as 4 h, remained elevated on day 1 and then decreased on days 3 and 7, but was still higher than those of saline groups. We also observed that Mp increased IL-17 protein levels in BAL fluid (Fig. 2). IL-17 was not detectable in any saline-treated mice and Mp-infected mice at 4 h. However, IL-17 protein levels were increased significantly in Mp-infected mice on day 1, and then decreased on days 3 and 7, which were still significantly higher than those of saline groups.
Fig. 2.
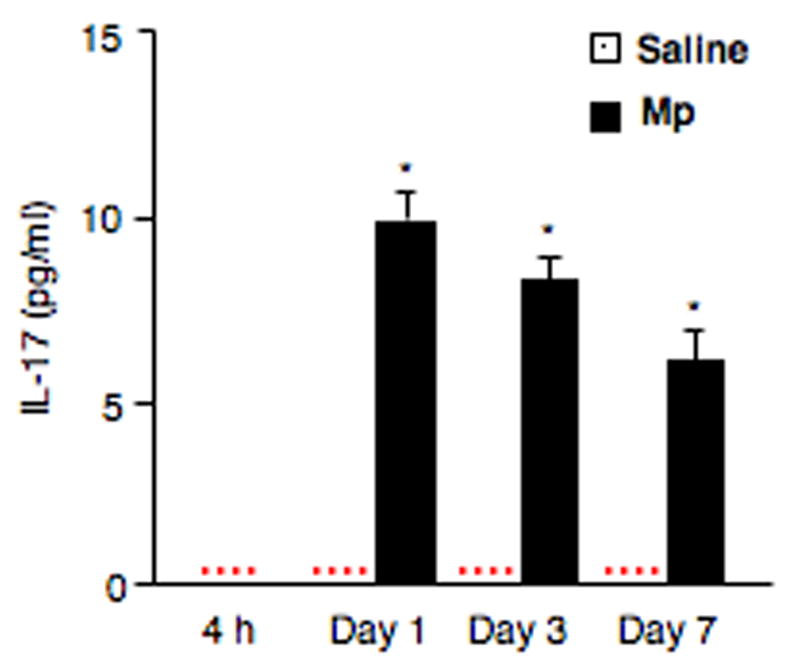
IL-17 protein levels in BAL fluid. Samples were collected from saline-treated (n=5/group) and M. pneumoniae (Mp)-infected mice (n=10/group) at 4 h, days 1, 3 and 7. Data are expressed as means ± SEM. * p < 0.01, matched-pairs t test between saline group and Mp group. Dotted line = undetectable level.
3.4. IL-23 induces expression of IL-17/IL-17F in pulmonary CD4+ T cells
We observed that IL-23 induced IL-17 production in cultured pulmonary CD4+ T cells in a dose- and time-dependent manner. It appears that 10 ng/ml of rmIL-23 was the lowest concentration required in inducing a peak production of IL-17. Therefore, for the remaining cell cultures, IL-23 was used at 10 ng/ml.
As shown in Figure 3A, in the absence of IL-23, CD4+ T cells from naïve mice did not produce IL-17 protein. However, CD4+ T cells from Mp-infected mice released detectable levels of IL-17 protein, especially those from the day 1 group. In the presence of IL-23, IL-17 protein levels in CD4+ T cells from naïve mice at all examined time points increased after 24 h and 48 h, and interestingly, they were similar to the levels in CD4+ T cells from Mp-infected mice without IL-23 stimulation. In addition, CD4+ T cells from Mp-infected mice further responded to IL-23 by enhancing IL-17 protein production. It is worth noting that with IL-23 stimulation the absolute IL-17 protein levels in CD4+ T cells from Mp-infected mice were 3 to 4-fold higher than those in CD4+ T cells from naïve mice.
Fig. 3.
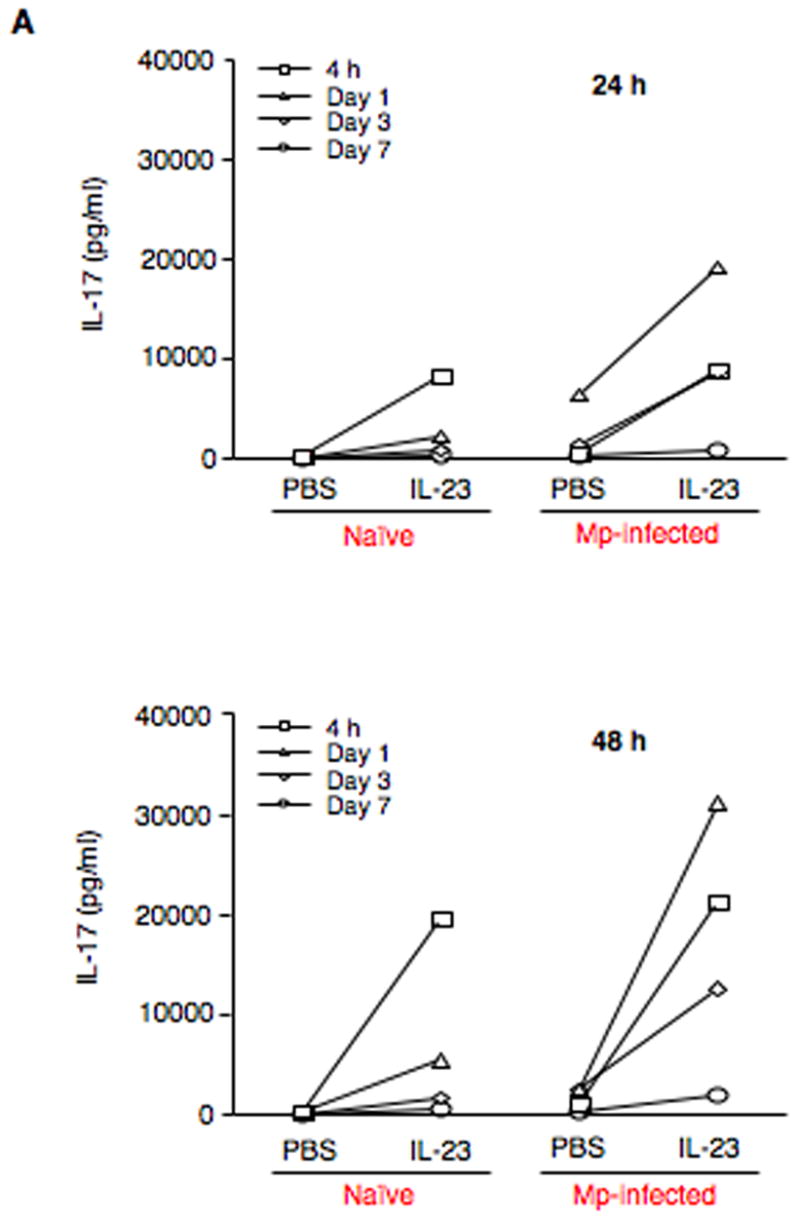
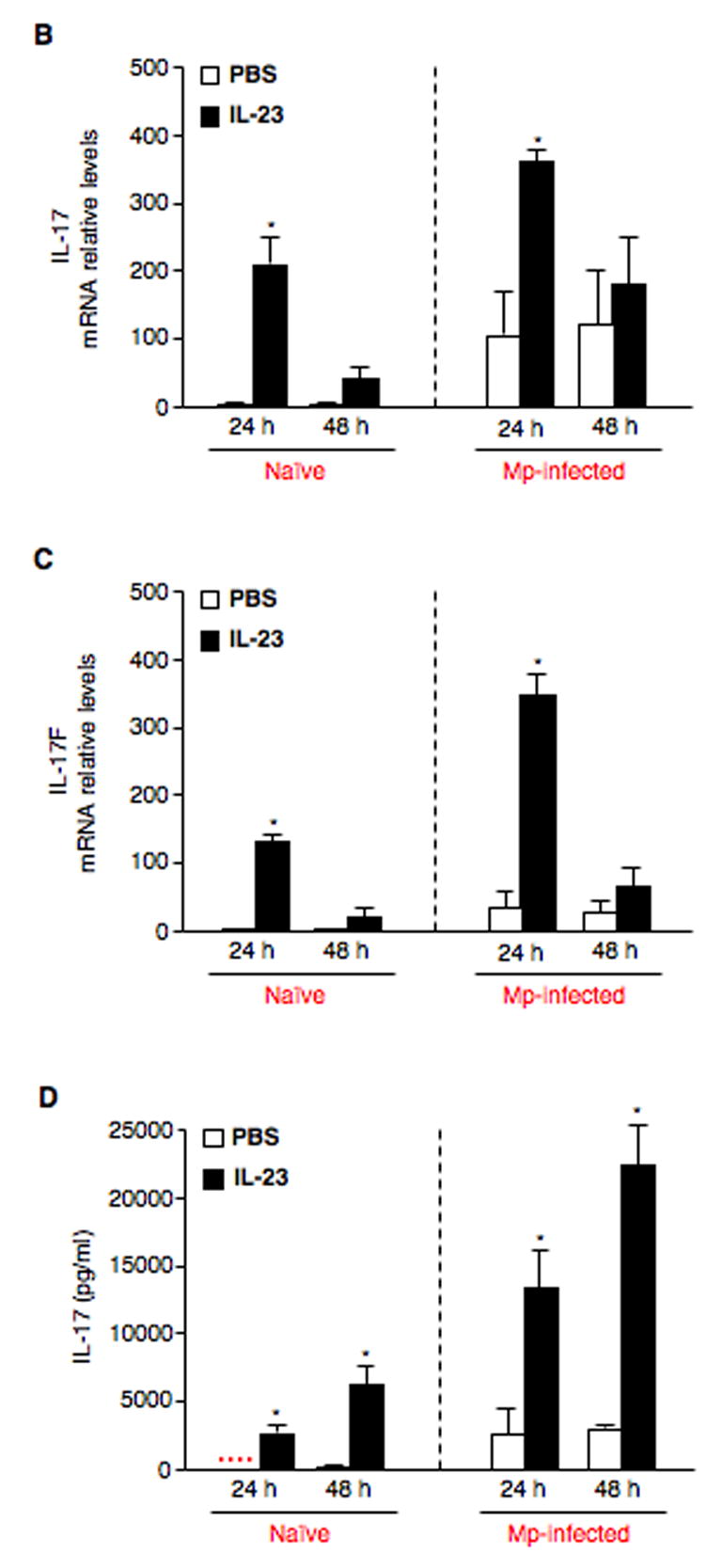
IL-23 increases expression of IL-17 family members by pulmonary CD4+ T cells. (A) Pulmonary CD4+ T cells isolated from naïve (saline-treated) and Mp-infected mice at 4 h, days 1, 3 and 7 were incubated in the absence (PBS) or presence of 10 ng/ml rmIL-23 for 24 h or 48 h. IL-17 protein in cell supernatants was measured by ELISA. To further confirm the findings in Fig. 3A, pulmonary CD4+ T cells from naïve (saline-treated) and Mp-treated mice at 24 h were incubated in the absence (PBS) or presence of 10 ng/ml rmIL-23 for 24 h or 48 h. Relative mRNA levels of IL-17 (B) and IL-17F (C), and protein levels of IL-17 (D) are expressed as means ± SEM from three independent replicates. *p < 0.05, matched-pairs t test comparing PBS with IL-23 treatment. Dotted line = undetectable level.
Based on our results shown in Fig. 3A, three more independent experiments were performed using pulmonary CD4+ T cells from naïve (saline-treated) and Mp-treated mice at 24 h to further confirm the effects of IL-23 on IL-17 and its family members. In the absence of IL-23, IL-17 mRNA levels were very low in naïve CD4+ T cells. IL-17 mRNA levels were significantly increased after IL-23 stimulation, especially at 24 h. Interestingly, even without in vitro IL-23 stimulation, CD4+ T cells from Mp-infected mice demonstrated higher levels of IL-17 mRNA than those of naïve CD4+ cells. With further IL-23 stimulation, IL-17 mRNA levels were significantly up-regulated at both time points, especially at 24 h (Fig. 3B). In addition, IL-17F mRNA levels in both naïve and Mp-infected CD4+ T cells showed a similar response pattern to IL-17 mRNA in the absence or presence of IL-23 (Fig. 3C). However, IL-23 did not significantly change IL-17C mRNA levels in both naïve and Mp-infected CD4+ T cells (data not shown). IL-17 protein levels were also measured in cell supernatants (Fig. 3D), which were essentially the same as the IL-17 mRNA data. Since IL-17F and IL-17C ELISA kits are not available, their protein levels could not be measured.
3.5. Effects of in vivo IL-23 neutralization
To examine whether blockade of IL-23 signaling would reduce Mp-induced expression of IL-17 family members and neutrophil function, a blocking experiment was performed. Anti-IL-23p19 Ab led to a 54% of reduction in BAL IL-17 protein levels compared with isotype antibody controls (p < 0.05). Furthermore, blocking both p19 and p40 subunits almost abolished Mp-induced IL-17 in BAL fluid (p < 0.01) (Fig. 4A). Similar pattern of reduced IL-17 and IL-17F mRNA levels in lung tissues was also found (Fig. 4B and 4C). However, unlike IL-17 and IL-17F, Mp-induced IL-17C mRNA levels were not reduced by p19 neutralizing Ab alone or in combination with p40-neutralizing Ab (data not shown).
Fig. 4.
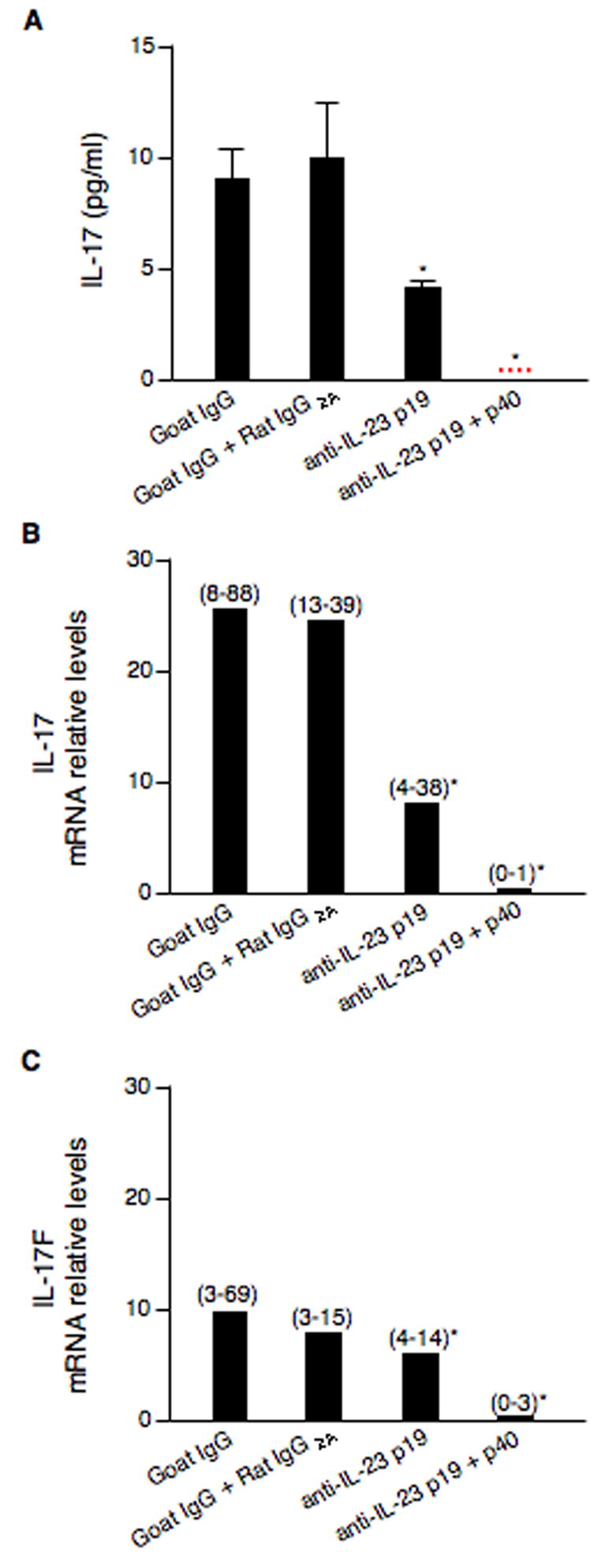
In vivo neutralization of IL-23 decreases expression of IL-17 family members. BALB/c mice (n=5–8/group) were intranasally inoculated with goat anti-mouse IL-23p19 Ab, rat anti-mouse IL-23/IL-12p40 Ab, goat IgG (control for anti-IL-23p19 Ab) or rat IgG2A (control for anti-IL-23/IL-12p40 Ab) for 2 h prior to a 50 μl of M. pneumoniae (107 CFUs/mouse) inoculation. Mice were sacrificed at 24 h post infection. Neutralization of IL-23p19 alone or in combination with the p40 subunit results in a significant reduction in: (A) IL-17 protein levels in BAL fluid; (B) lung tissue IL-17 mRNA levels, and (C) lung tissue IL-17F mRNA levels. Data are expressed as means ± SEM from two independent replicates. *p < 0.05, matched-pairs t test comparing specific IL-23 neutralizing antibody treatment with isotype antibody treatment. Dotted line = undetectable level.
To determine the functional consequences of blocking IL-23 signaling, we measured in vivo bacterial clearance by culturing Mp in lung tissues and lung neutrophil activity. Compared with isotype antibody controls, lung Mp levels at 24 h post infection were 50% and 70% higher in mice treated with p19 neutralizing Ab alone (p = 0.16) or in combination with p40-neutralizing Ab (p = 0.13), respectively (Fig. 5A). Neutralizing p19 had a minimal effect on MPO activity in lungs (a 10% decrease) as compared to the isotype antibody (goat IgG) control. However, blocking both p19 and p40 subunits significantly reduced lung MPO activity (a 43% decrease) as compared to the isotype antibody (goat IgG + Rat IgG2A) control (p < 0.01) (Fig. 5B). We next measured the released MPO activity by neutrophils in BAL. MPO activity (mU) per 106 neutrophils was lower in mice treated with p19 neutralizing Ab alone (a 33% decrease, p = 0.44) or in combination with p40-neutralizing Ab (a 60% decrease, p = 0.19) as compared to the relevant isotype antibody control (Fig. 5C).
Fig. 5.
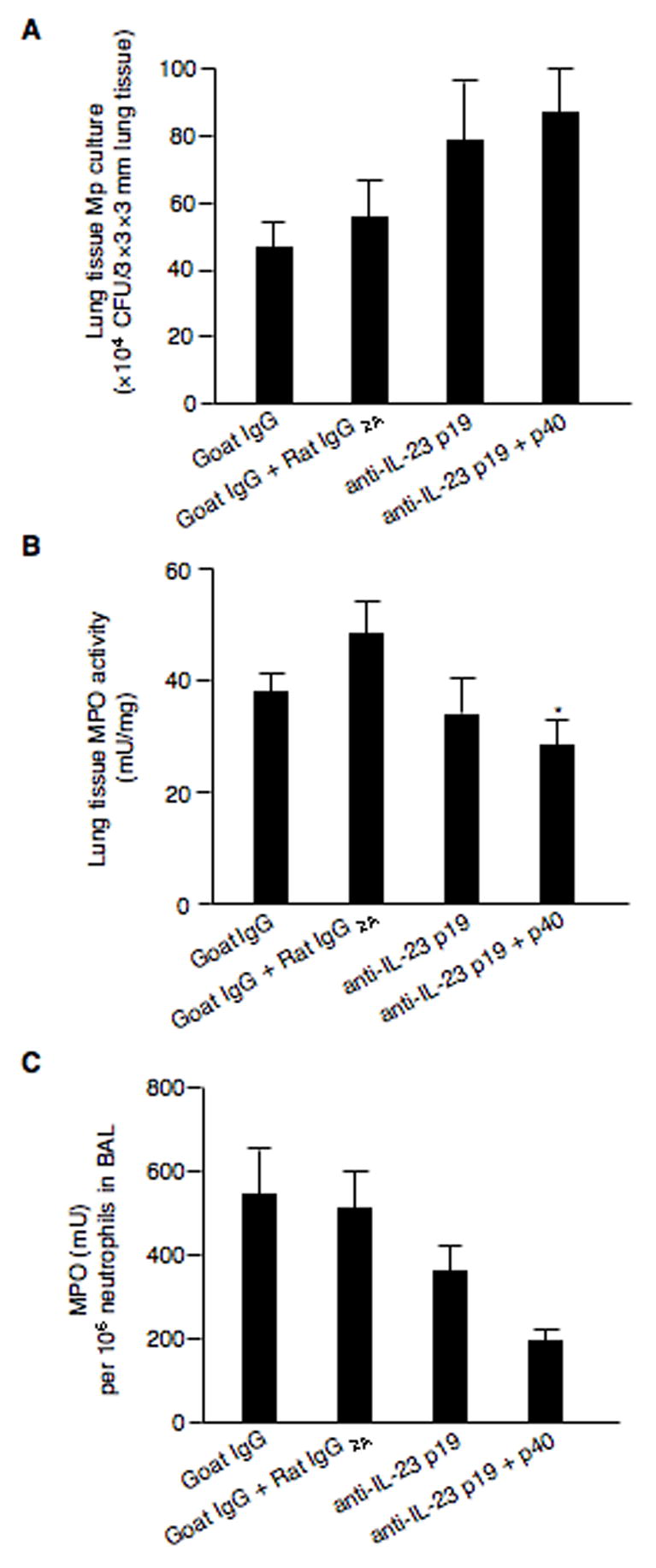
In vivo neutralization of IL-23 dampens bacterial clearance and neutrophil activity. BALB/c mice (n=5–8/group) were intranasally inoculated with goat anti-mouse IL-23p19 Ab, rat anti-mouse IL-23/IL-12p40 Ab, goat IgG (control for anti-IL-23p19 Ab) or rat IgG2A (control for anti-IL-23/IL-12p40 Ab) for 2 h prior to a 50 μl of M. pneumoniae (107 CFUs/mouse) inoculation. Mice were sacrificed at 24 h post infection. M. pneumoniae culture of lung tissues (A), MPO activity in lungs (B), and MPO activity levels normalized by neutrophil numbers in BAL (C). Data are expressed as means ± SEM from two independent replicates. *p < 0.01, matched-pairs t test comparing specific IL-23 neutralizing antibody treatment with isotype antibody treatment.
4. Discussion
This is the first study to reveal the role of IL-23/IL-17 axis in host defense against the respiratory M. pneumoniae infection.
In the present study, Mp was found to significantly up-regulate lung IL-23p19 mRNA as early as 4 h after the infection in mice. IL-23 is shown to be produced by APCs (e.g. activated macrophages and dendritic cells) and participates in the regulation of T cell responses [13, 21]. Although alveolar macrophages have been proposed as the very first line of host innate immune responses to environmental insults such as M. pneumoniae infection, little is known about the pathways involved [22]. Here, we have also shown noticeable increased levels of IL-23p19 mRNA in pAMs ex vivo (freshly isolated pAMs from Mp-infected mice) as well as in vitro (naïve pAMs infected with Mp). These data suggest an important role of AMs through an enhanced production of IL-23 in host defense against Mp.
IL-23 has recently been described as a proximal regulator to preferentially i nduce production of IL-17/IL-17F by T cells from human cord blood or mouse spleens [10–12]. Although activated CD4+ T cells are believed to a major source of IL-17, activated CD8+ T cells, neutrophils and eosinophils are also able to produce IL-17 [11, 23–25]. Since activation of CD4+ T cells has been shown to be a key feature in lungs with Mp infection, we chose to study the effects of IL-23 on IL-17 family members in isolated pulmonary CD4+ T cells from naïve (saline treatment) and Mp-infected mice. In the absence of IL-23, in contrast to the naïve ones, pulmonary CD4+ T cells from Mp-infected mice produced detectable levels of IL-17 protein, which were further enhanced upon exogenous IL-23 treatment. Importantly, our current study extended previous studies in that IL-23 was also shown to increase IL-17F expression in pulmonary CD4+ T cells from both naïve and especially Mp-infected mice. However, unlike their robust production of IL-17 and IL-17F, pulmonary CD4+ T cells represent a negligible source of IL-17C. These results have further clarified the role of IL-23 in regulating IL-17 family members in respiratory Mp infection. Whether our findings in the present study are applicable to other respiratory bacterial infections awaits future studies.
The pAMs and pulmonary CD4+ T cell culture data were further supported by the in vivo data collected from Mp-infected mice. Mp infection increased lung IL-23, IL-17 and IL-17F mRNA levels as early as 4 h, which were remained until day 7. Thus, our study has expanded the list of pathogens (mycoplasma in addition to Gram-positive and Gram-negative bacteria) that may utilize IL-23/IL-17 axis to initiate a robust host defense against the invading microorganisms [26–30].
Additional pieces of evidence for a pivotal role of IL-23/IL-17 axis in host defense against Mp infection come from our experiments in which IL-23 neutralizing antibodies were utilized to examine the impacts of a deficient IL-23 signaling on the downstream immune responses to the Mp infection. Blocking p19 subunit alone partly (30%–70%) inhibited Mp-induced IL-17 protein production in BAL and IL-17/IL-17F mRNA expression in lung tissues. Moreover, blocking both p19 and p40 subunits resulted in an abrogation of Mp-induced IL-17 protein and IL-17/IL-17F mRNA expression. These in vivo data indicate that IL-23 is essential in regulation of IL-17 family members production/expression in response to Mp infection. The consequences of a defective IL-23/IL-17 axis in host defense against Mp remain unclear. Here, a trend toward an increased lung Mp load, lower neutrophil content in lung tissue as well as in BAL reflected by MPO activity levels was observed in mice with IL-23 signaling being blocked. As a significant correlation between tissue MPO levels and neutrophil accumulation is well documented [20, 31–35], the decreased MPO activity levels due to IL-23 blocking suggest less neutrophil accumulation in the lung with reduced IL-17/IL-17F. In addition, although an abrogation of Mp-induced IL-17 /IL-17F and significantly decreased neutrophil levels were seen in the lung after IL-23 blocking, there was only a good, but not significant trend toward an increase of lung Mp load in vivo. We realize that IL-23-induced-IL-17 acts as a very important, but not a sole factor contributing to Mp clearance. Many other factors may also be involved in Mp clearance in our acute infection model. These may include innate immune cells (e.g., macrophages), antimicrobial substances from epithelial cells and macrophages, and other cytokines or chemokines (e.g., IL-18, IFN-γ and KC). Collectively, our data suggest that IL-17/IL-17F production in lung defense against Mp is IL-23 dependent. IL-17 and IL-17F production may serve as a bridge linking the activation of T lymphocytes to the sustained accumulation and activity of lung neutrophils. Decreased IL-17/IL-17F production due to a deficient IL-23 signaling contributes to dampen the lung neutrophil activity and consequently delays Mp clearance from the lung.
Unlike the relatively well-studied IL-17 and IL-17F, the potential role of other IL-17 family members in host defense against bacterial infections remains much unknown. In the present study, we found that lung IL-17C mRNA levels were increased in response to an in vivo Mp infection in a time-dependent manner. However, both pulmonary CD4+ T cell culture experiments and in vivo IL-23 blocking showed that IL-23 did not have any significant regulatory effect on IL-17C mRNA expression. Our novel data suggest that IL-17C may utilize an alternative upstream regulatory pathway that increases IL-17C production in host inflammatory process in a M. pneumoniae infection.
In conclusion, IL-23 was shown to be inducible in alveolar macrophages in response to M. pneumoniae infection. Production of IL-17 and IL-17F by pulmonary CD4+ T cells is IL-23 dependent and may act as an interface between innate and adaptive immunity in lung defense (e.g., neutrophil recruitment and activation) against M. pneumoniae. Our present study has significantly extended previous results in many encapsulated bacterial infections by demonstrating that IL-23/IL-17s axis is also an important lung defense mechanism involved in an M. pneumoniae infection. These research findings will further improve our understanding of cytokine regulatory mechanisms underlying the M. pneumoniae infection in lung diseases associated with neutrophil accumulation.
Acknowledgments
This work was supported by National Institutes of Health (grant PO1 HL073907) and American Lung Association-Colorado. We thank Dr. Ronald J. Harbeck for helpful suggestions and Taylor Moss, Samithamby Jeyasseelan, Terri Okotoghaide, Jeff Zdunek and Clare M. Ennis for technical assistance.
Abbreviations
- BAL
bronchoalveolar lavage
- CFUs
colony forming units
- Mp
Mycoplasma pneumoniae
- MPO
myeloperioxidase
- pAMs
primary alveolar macrophages
- rm
recombinant murine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilson MH, Collier AM. Ultrastructural study of Mycoplasma pneumoniae in organ culture. J Bacteriol. 1976;125:332–339. doi: 10.1128/jb.125.1.332-339.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin RJ, Kraft M, Chu HW, Berns EA, Cassell GH. A link between chronic asthma and chronic infection. J Allergy Clin Immunol. 2001;107:595–601. doi: 10.1067/mai.2001.113563. [DOI] [PubMed] [Google Scholar]
- 3.Lieberman D, Lieberman D, Ben-Yaakov M, Lazarovich Z, Hoffman S, Ohana B, Friedman MG, Dvoskin B, Leinonen M, Boldur I. Infectious etiologies in acute exacerbation of COPD. Diagn Microbiol Infec Dis. 2001;40:95–102. doi: 10.1016/s0732-8893(01)00255-3. [DOI] [PubMed] [Google Scholar]
- 4.Kraft M, Cassell GH, Henson JE, Watson H, Williamson J, Marmion BP, Gaydos CA, Martin RJ. Detection of Mycoplasma pneumoniae in the airways of adults with chronic asthma. Am J Respir Crit Care Med. 1998;158:998–1001. doi: 10.1164/ajrccm.158.3.9711092. [DOI] [PubMed] [Google Scholar]
- 5.Martin RJ, Chu HW, Honour JM, Harbeck RJ. Airway inflammation and bronchial hyperresponsiveness after Mycoplasma pneumoniae infection in a murine model. Am J Respir Crit Care Med. 2001;24:577–582. doi: 10.1165/ajrcmb.24.5.4315. [DOI] [PubMed] [Google Scholar]
- 6.Chu HW, Breed R, Rino JG, Harbeck RJ, Sills MR, Martin RJ. Repeated respiratory Mycoplasma pneumoniae infections in mice: effect of host genetic background. Microbes Infect. 2006 doi: 10.1016/j.micinf.2006.02.014. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 7.Chan ED, Welsh CH. Fulminant Mycoplasma pneumoniae pneumonia. West J Med. 1995;162:133–142. [PMC free article] [PubMed] [Google Scholar]
- 8.Opitz O, Pietsch K, Ehlers S, Jacobs E. Cytokine gene expression in immune mice reinfected with Mycoplasma pneumoniae: the role of T cell subsets in aggravating the inflammatory response. Immunobiology. 1996;196:575–87. doi: 10.1016/s0171-2985(97)80073-3. [DOI] [PubMed] [Google Scholar]
- 9.Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol. 1993;150:5445–5456. [PubMed] [Google Scholar]
- 10.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 11.Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ, Shellito JE, Schurr JR, Bagby GJ, Nelson S, Kolls JK. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J Immunol. 2003;170:4432–4436. doi: 10.4049/jimmunol.170.9.4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vanden Eijnden S, Goriely S, De Wit D, Willems F, Goldman M. IL-23 up-regulates IL-10 and induces IL-17 synthesis by polyclonally activated naive T cells in human. Eur J Immunol. 2005;35:469–475. doi: 10.1002/eji.200425677. [DOI] [PubMed] [Google Scholar]
- 13.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 14.Hoeve MA, Savage ND, de Boer T, Langenberg DM, de Waal Malefyt R, Ottenhoff TH, Verreck FA. Divergent effects of IL-12 and IL-23 on the production of IL-17 by human T cells. Eur J Immunol. 2006;36:661–670. doi: 10.1002/eji.200535239. [DOI] [PubMed] [Google Scholar]
- 15.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly MN, Kolls JK, Happel K, Schwartzman JD, Schwarzenberger P, Combe C, Moretto M, Khan IA. Interleukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against Toxoplasma gondii infection. Infect Immun. 2005;73:617–621. doi: 10.1128/IAI.73.1.617-621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleinschek MA, Muller U, Brodie SJ, Stenzel W, Kohler G, Blumenschein WM, Straubinger RK, McClanahan T, Kastelein RA, Alber G. IL-23 enhances the inflammatory cell response in Cryptococcus neoformans infection and induces a cytokine pattern distinct from IL-12. J Immunol. 2006;176:1098–1106. doi: 10.4049/jimmunol.176.2.1098. [DOI] [PubMed] [Google Scholar]
- 18.Lahn M, Kanehiro A, Takeda K, Terry J, Hahn YS, Aydintug MK, Konowal A, Ikuta K, O'Brien RL, Gelfand EW, Born WK. MHC class I-dependent Vgamma4+ pulmonary T cells regulate alpha beta T cell-independent airway responsiveness. Proc Natl Acad Sci USA. 2002;99:8850–8855. doi: 10.1073/pnas.132519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 20.Jeyaseelan S, Chu HW, Young SK, Worthen GS. Transcriptional profiling of lipopolysaccharide-induced acute lung injury. Infect Immun. 2004;72:7247–7256. doi: 10.1128/IAI.72.12.7247-7256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19:641–644. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 22.Hickman-Davis JM, Lindsey JR, Zhu S, Matalon S. Surfactant protein A mediates mycoplasmacidal activity of alveolar macrophages. Am J Physiol. 1998;274:L270–277. doi: 10.1152/ajplung.1998.274.2.L270. [DOI] [PubMed] [Google Scholar]
- 23.Yang D, Chen Q, Hoover DM, Staley P, Tucker KD, Lubkowski J, Oppenheim JJ. Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J Leukoc Bio. 2003;74:448–455. doi: 10.1189/jlb.0103024. [DOI] [PubMed] [Google Scholar]
- 24.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, Schwarzenberger P, Shellito JE, Kolls JK. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am J Respir Cell Mol Biol. 2001;25:335–340. doi: 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- 26.Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. 2003;170:2106–2112. doi: 10.4049/jimmunol.170.4.2106. [DOI] [PubMed] [Google Scholar]
- 27.Miyamoto M, Prause O, Sjostrand M, Laan M, Lotvall J, Linden A. Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J Immunol. 2003;170:4665–4672. doi: 10.4049/jimmunol.170.9.4665. [DOI] [PubMed] [Google Scholar]
- 28.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107–6115. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 29.Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwarzenberger P, La Russa V, Miller A, Ye P, Huang W, Zieske A, Nelson S, Bagby GJ, Stoltz D, Mynatt RL, Spriggs M, Kolls JK. IL-17 stimulates granulopoiesis in mice: use of an alternate, novel gene therapy-derived method for in vivo evaluation of cytokines. J Immunol. 1998;161:6383–6389. [PubMed] [Google Scholar]
- 31.Castillo MJ, Nakajima K, Zimmerman M, Powers JC. Sensitive substrates for human leukocyte and porcine pancreatic elastase: a study of the merits of various chromophoric and fluorogenic leaving groups in assays for serine proteases. Anal Biochem. 1979;99:53–64. doi: 10.1016/0003-2697(79)90043-5. [DOI] [PubMed] [Google Scholar]
- 32.Morikawa K, Kikuchi Y, Abe S, Yamazaki M, Mizuno D. Early cellular responses in the peritoneal cavity of mice to antitumor immunomodulators. Gann. 1984;75:370–378. [PubMed] [Google Scholar]
- 33.Schmekel B, Karlsson SE, Linden M, Sundstrom C, Tegner H, Venge P. Myeloperoxidase in human lung lavage. I. A marker of local neutrophil activity. Inflammation. 1990;14:447–454. doi: 10.1007/BF00914095. [DOI] [PubMed] [Google Scholar]
- 34.Katiyar SK, Mukhtar H. Green tea polyphenol (−)-epigallocatechin-3-gallate treatment to mouse skin prevents UVB-induced infiltration of leukocytes, depletion of antigen-presenting cells, and oxidative stress. J Leukoc Biol. 2001;69:719–726. [PubMed] [Google Scholar]
- 35.Lee JS, Frevert CW, Matute-Bello G, Wurfel MM, Wong VA, Lin SM, Ruzinski J, Mongovin S, Goodman RB, Martin TR. TLR-4 pathway mediates the inflammatory response but not bacterial elimination in E. coli pneumonia. Am J Physiol Lung Cell Mol Physiol. 2005;289:L731–738. doi: 10.1152/ajplung.00196.2005. [DOI] [PubMed] [Google Scholar]