

Abstract
Both neointimal hyperplasia and inward remodeling contribute to restenosis and lumen loss. Nogo-B has been recently described as an inhibitor of vascular injury and neointimal hyperplasia. To determine whether Nogo-B expression may be a mediator of inward remodeling, we examine the localization of expression of Nogo-B in an in vivo model that examines both neointimal hyperplasia and inward remodeling. The rabbit carotid artery was subjected to balloon injury, outflow branch ligation to reduce flow, or both balloon injury and reduction in flow. In balloon injury-induced neointimal hyperplasia Nogo-B expression was reduced in the intima and media but stimulated in the adventitia. In low flow-induced inward remodeling medial Nogo-B expression was not reduced and adventitial Nogo-B expression was not stimulated. Low flow significantly augmented balloon injury-induced neointimal hyperplasia and was accompanied by reduced intimal and medial Nogo-B expression, and increased adventitial Nogo-B expression in both smooth muscle cells and macrophages. Low flow-induced inward remodeling is not associated with changes in medial Nogo-B expression and is distinct from injury-induced neointimal hyperplasia. Pharmacological strategies to inhibit neointimal hyperplasia and restenosis using normal flow models may only partially account for lumen loss and therefore may not accurately predict responses in patients with extensive outflow disease.
Keywords: neointimal hyperplasia, flow-induced remodeling, Nogo-B, rabbit
1. Introduction
Restenosis following arterial injury may be mediated by both neointimal hyperplasia as well as inward remodeling (Kuntz, Gibson et al. 1993). The immediate response to injury induces an cascade of cytokine and inflammatory mediators from platelets, endothelial and smooth muscle cells, stimulating vascular smooth muscle cell proliferation and migration, resulting in the space-occupying lesion of neointimal hyperplasia and lumen loss (Austin, Ratliff et al. 1985; Bauters and Isner 1997; Schwartz 1998; Ohashi, Matsumori et al. 2000; Rectenwald, Moldawer et al. 2000). Inward vessel remodeling is an endothelial-dependent compensatory response to chronic low blood flow and shear stress (Langille and O'Donnell 1986; Zarins, Zatina et al. 1987; Tronc, Wassef et al. 1996; Qin, Dardik et al. 2001; Tuttle, Nachreiner et al. 2001); multiple studies suggest that inward remodeling is also a significant factor contributing to the lumen compromise of restenosis (Kamiya and Togawa 1980; Kohler and Jawien 1992; Kakuta, Currier et al. 1994; Post, Borst et al. 1994; Lafont, Guzman et al. 1995; Harmon, Couper et al. 2000), and must be treated successfully to address restenosis (Post, de Smet et al. 1997). The ability of an injured vessel to normalize shear stress may be an important initiating factor stimulating inward remodeling (Post, Borst et al. 1995; Girerd, London et al. 1996; Guzman, Abe et al. 1997), and identifies the important role of shear stress in vascular remodeling (Krams, Wentzel et al. 1998; Song, Kocharyan et al. 2000; Ward, Tsao et al. 2001; Paszkowiak and Dardik 2003; Ward, Tsao et al. 2003; Wentzel, Gijsen et al. 2003; Hanratty, Murrell et al. 2004).
Nogo-B has been recently demonstrated to be expressed in both endothelial cells and smooth muscle cells, and regulate vascular remodeling in vivo (Acevedo, Yu et al. 2004). In particular, loss of Nogo-B from the vessel media correlates with neointimal expansion and lumen loss; adenoviral-mediated gene transfer of Nogo-B in Nogo-B knockout mice rescues injury induced neointimal expansion (Acevedo, Yu et al. 2004). It is not currently known whether Nogo-B expression changes with low shear stress and resultant inward remodeling. To determine whether Nogo-B expression changes during low flow-induced inward remodeling, as well as low flow-augmented neointimal hyperplasia, we examined the distribution of Nogo-B expression in the different layers in an injured vessel, and whether this distribution was modified by low shear stress. In addition, we determined whether Nogo-B was expressed in smooth muscle cells or macrophages in balloon injured vessels.
2. Methods
2.1. Animal model
Adult male New Zealand white rabbits (3.0 ± 0.2 kg) were used for all procedures. Rabbits were allowed to acclimate to the facility and maintained on a light/dark cycle of 12/12 hours at 24°C; access to a normal diet and water was allowed as desired. Animals were weighed and checked for signs of infection daily. Animal care complied with “Principles of Laboratory Animal Care” and the “Guide for the Care and Use of Laboratory Animals” (NIH Publication, revised 1996).
Rabbits were anesthetized with an intramuscular injection of ketamine (50 mg/kg), xylazine (5 mg/kg), and acepromazine (1 mg/kg) and placed on a warming blanket. Vital signs including oxygen saturation were monitored continually. Neither mechanical ventilation nor supplemental intravenous fluid administration was required. No prophylactic antibiotics were given.
After preparing the neck in sterile fashion with alcohol and betadine, a vertical midline incision was made and the right carotid artery was exposed. Animals in experimental groups received either balloon injury (B) to the right common carotid artery, flow reduction (low flow, LF), or both balloon injury and flow reduction (B+LF); control animals had no additional intervention performed.
Balloon injury was performed by introducing a 2-Fr balloon catheter via a distal branch of the external carotid artery and passing the catheter in retrograde fashion into the proximal common carotid artery; the balloon was inflated and passed through the entire common carotid artery three times (approximately 4 cm). After the third pass the catheter was removed and the branch vessel ligated. Flow in the right common carotid artery was reduced by ligation of the distal outflow branches of the common carotid artery other than the internal carotid artery; reduction in flow was verified with a Doppler flow probe (Transonic Systems Inc, Ithaca, NY). In rabbits receiving both balloon injury and flow reduction, the balloon injury was performed prior to ligation of the distal branches.
Neither heparin nor antiplatelet agents were given. Blood flow was measured with a Doppler flow probe and recorded prior to and after intervention in all animals. The external diameter of the common carotid artery was measured with a micrometer at its midpoint, two centimeters from the bifurcation. The incision was closed in two layers and the rabbits were placed under a warming light until fully recovered from anesthesia. After recovery, rabbits were allowed access to a normal diet and water as desired.
2.2. Specimen harvest
Animals were anesthetized 7 or 21 days after the first procedure. 5-bromo-2′-deoxy-uridine (BrdU) (30 mg/kg, intraperitoneally) was given 24 and 12 hours prior to specimen harvest in animals examined on postoperative day 7. The previously made incision was opened and both common carotid arteries were examined; flow was measured with a Doppler probe and vessel external diameter directly recorded. The vessels were flushed with saline and then perfusion fixed in-situ with 500cc 1% formalin at a perfusion pressure of 100–110 mmHg; the arteries were harvested en bloc for further immersion fixation in 10% formalin. Animals were allowed to exsanguinate under anesthesia.
2.3. Morphology and Analysis
Vessels were examined at the midpoint of the common carotid artery and stained with hematoxylin and eosin for computer-aided morphometry (see below). Shear stress was calculated from the Poiseuille formula SS = 4μQ/πr3 where μ is the viscosity of blood, Q is the blood flow, and r is the vessel radius. The vessel radius was estimated to be half the external measured diameter; the viscosity of blood is approximately 0.035 poise. Shear stress was calculated prior to and immediately after the initial procedure, as well as on day 21 immediately prior to vessel harvest.
For BrdU detection, sections were incubated with primary anti-BrdU antibody (BD Pharmingen, Franklin Lakes, NJ) for 2hours at room temperature prior to incubation with 3,3′-diaminobenzidine tetrahydrochloride (Roche). The sections were counterstained with Mayer’s Hematoxylin. Positively- and negatively-staining nuclei were directly counted.
Apoptotic cells were detected using the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) method using the In Situ Apoptosis Detection Kit (Chemicon) according to the manufacturer's instructions. Tissue sections were counterstained with hematoxylin. Apoptotic bodies stained brown, whereas nonapoptotic nuclei were visualized as blue. The number of the TUNEL-positive cells was defined as the percentage of TUNEL-positive cells per total number of cells in each layer.
Immunohistochemistry was performed in post-operative day 7 specimens. 5 μm thick slides were sectioned from specimens embedded in frozen OCT, fixed with cold acetone and rinsed in PBS. Endogenous peroxidase activity was blocked by 10 min incubation in 3% H2O2 in methanol at room temperature. The slides were then rinsed once more with PBS. For Nogo-B expression the slides were bathed with goat polyclonal IgG antibody Anti-Nogo (Santa Cruz Biotechnologies, Santa Cruz, CA; Nogo (N-18), #sc-11027, in PBS, dilution 1:3000) in a moist chamber overnight at 4°C. The slides were subsequently rinsed in PBS and treated with the secondary antibody (Santa Cruz Biotechnologies donkey anti-goat IgG-B, #sc-2042, in Dako Antibody Diluent with Background Reducing Components, dilution 1:200) at room temperature for 30 minutes. Following another wash with PBS, the sections were treated with Vectastain Elite ABC solution (Vector Laboratories, Burlingame, CA) for 30 minutes. Visualization was performed using Vector NovaRed substrate at room temperature for approximately 70 seconds. Sections were counterstained with hematoxylin, dehydrated, and mounted in Surgipath Sub-X mounting medium.
For immunofluorescence studies, sections were cut to 5μm thickness and stored at −20°C. Following fixation with cold acetone, endogenous peroxidase activity was quenched with 0.3% H2O2 in methanol. The first antibodies were then applied as follows: anti-Nogo-B goat polyclonal IgG (SantaCruz, 1:5000), monoclonal anti-smooth muscle alpha actin (Sigma, 1:500), monoclonal mouse anti-rabbit macrophage RAM-11 (Dako, 1:100). Alexa Fluor 488 and 568 were used for fluorescence. All samples were treated by Autofluorescent Eliminator Reagent (Chemicon, CA) and mounted by Vectashield hard set mounting medium with DAPI (Vector Laboratories, CA). Images were captured with an Axiovert 200M (Carl Zeiss MicroImaging, NY) under identical conditions. Cells staining positively for alpha actin or RAM-11 were directly counted.
Analysis was performed using the Metamorph Imaging System (Molecular Devices Corporation, Sunnyvale, CA). Each section was photographed at eight equidistant sampling points at a magnification of 20X. Using the Metamorph software the areas of the intima/neointima, media, and adventitia as well as cross-sectional area were selected for each photograph. The intima was defined as the area above the internal elastic lamina, in the luminal direction; the area containing neointimal SMC was included when present. The threshold for positive staining was set and analysis was performed by the Metamorph program designating the percent positive-stained area for each respective region. Each artery was assessed using three separate sections and in blinded fashion; initial results of specimen morphometry were confirmed with computerized digital morphometry (ImageJ 1.3, NIH).
2.4. Statistical analysis
Results are presented as mean ± SEM. Morphometric and hemodynamic results were analyzed using ANOVA and subgroups compared with Fischer’s PLSD post-hoc test (Statview 5.0, SAS Institute, Cary, NC). A p-value ≤ 0.05 was considered statistically significant.
3. Results
3.1. A reduction in blood flow augments neointimal thickening after balloon injury
Rabbits were sham operated (control, n=21), balloon injured (B), exposed to flow reduction (low flow, LF), or both procedures (B+LF) and several parameters measured. As seen in Figure 1, carotid arterial blood flow (Figure 1A) and shear stress (Figure 1B) were reduced after 21 days in the LF and B+LF groups. There were no differences in carotid artery external diameters before or after the procedures (data not shown), but external diameters were reduced after 21 days in the LF and B+LF groups (Figure 1C), suggesting inward remodeling in LF vessels. As seen in representative images in Figure 1D, treatment with B induced neointimal thickening (column 2, upper panel low magnification and lower panel high magnification) compared to control vessels (column 1, both panels); similarly, treatment with LF did not grossly affect vessel architecture (column 3, both panels). However, combining B with LF augmented the balloon injury-induced neointimal thickening (column 4, both panels). Morphometric analysis showed that lumen diameter was significantly reduced only in animals having both balloon injury and LF (Figure 1E). Medial areas were similar between the groups (Figure 1F) and the ratio of neointimal:medial thickness was increased after balloon injury; an effect augmented by combining balloon injury with LF (Figure 1G). These data show that LF enhances neointimal thickening formed in response to balloon injury as previously described (Song, Kocharyan et al. 2000; Ward, Tsao et al. 2001; Hanratty, Murrell et al. 2004).
Figure 1.
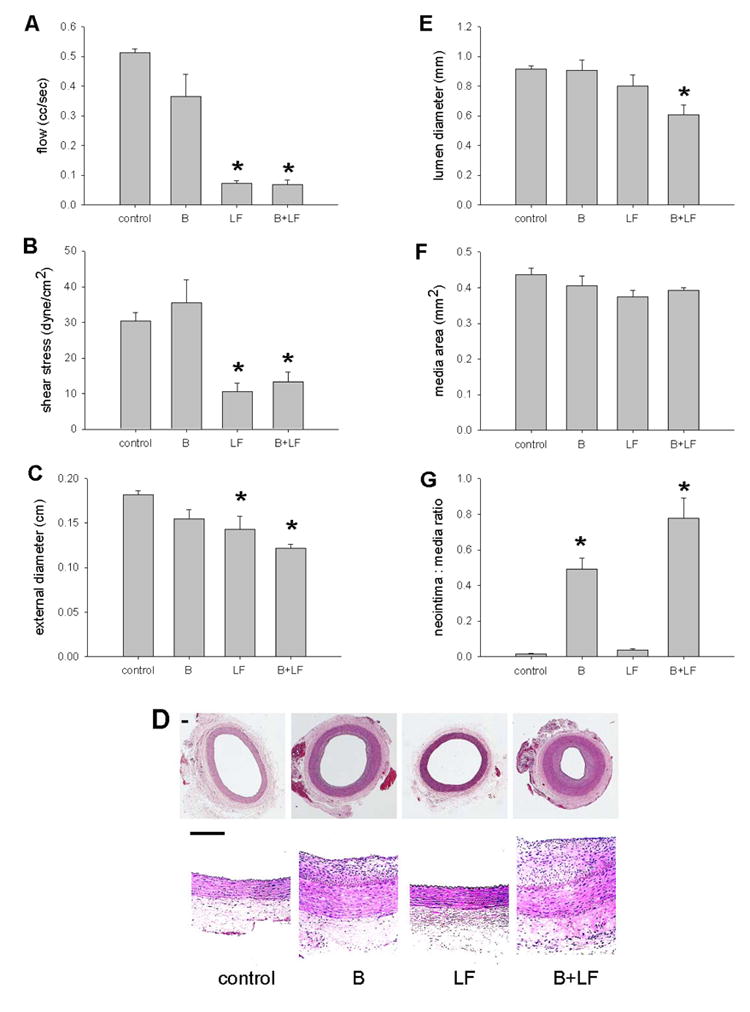
Effects of interactions on vessel size and hemodynamics (21d). A) Reduced flow in animals treated with low flow (LF) (*, p<.0001, post-hoc) or balloon injury and low flow (B + LF) (*, p<.0001) compared to control animals or animals treated with balloon injury (B) alone (p<.0001, ANOVA; n=21). B) Reduced shear stress in animals treated with LF (*, p=.01, post-hoc) or B+LF (*, p=.03) compared to control animals or animals treated with B alone (p=.001, ANOVA). C) Reduced diameter in animals treated with LF (*, p=.04, post-hoc) or B+LF (*, p=.003) compared to control animals or animals treated with B alone (p=.02, ANOVA). The 21% decrease in diameter in the LF group is similar to previous reports.(Langille and O'Donnell 1986) D) Representative photomicrographs of low and high power magnification of vessels (control, B, LF, B+LF). Scale bar, 100 μm. E) Reduced lumen diameter in animals treated with B+LF (*, p=.02, post-hoc) compared to control animals (p=.02, ANOVA). F) Similar media area between animal groups (p=.28, ANOVA). G) Increased neointima:media ratio in animals treated with B or B+LF (p<.0001, ANOVA). The increased neointima:media ratio in B+LF, compared to the increase in B, is significant (*, p=.01, post-hoc).
3.2. Flow-induced remodeling is distinct from vessel injury-evoked remodeling
To determine the rate of proliferation of cells within the wall in response to B, LF, or B+LF, proliferating cells were examined by injections of the DNA analog BrdU after 7 days of remodeling (n=12). In sham operated animals, there were no detectable BrdU-positive cells in the vessel wall. B increased the number of BrdU-positive cells in all layers of the vessel wall whereas LF only increased proliferating cells in the adventitia. Combining B+LF also increased the number of BrdU-positive cells in all layers of the vessel, with the majority of the proliferating cells in the media (Figure 2A). To examine the relative levels of apoptosis, TUNEL staining was performed. In control vessels, TUNEL-positive cells were undetectable. B increased the number TUNEL-positive cells uniformly in all layers of the vessel examined whereas LF only increased TUNEL-positive cells in the adventitial layer of vessels. Combining B+LF increased TUNEL-positive cells uniformly in all vessel layers, with the majority of in the TUNEL positive cells uniformly throughout the media (Figure 2B). These results suggest that B stimulates both proliferation and apoptosis in all vessel layers, but LF stimulates apoptosis and proliferation predominantly in the adventitia, and the combination of B+LF further stimulates cell turnover in all vessel layers but predominantly in media.
Figure 2.
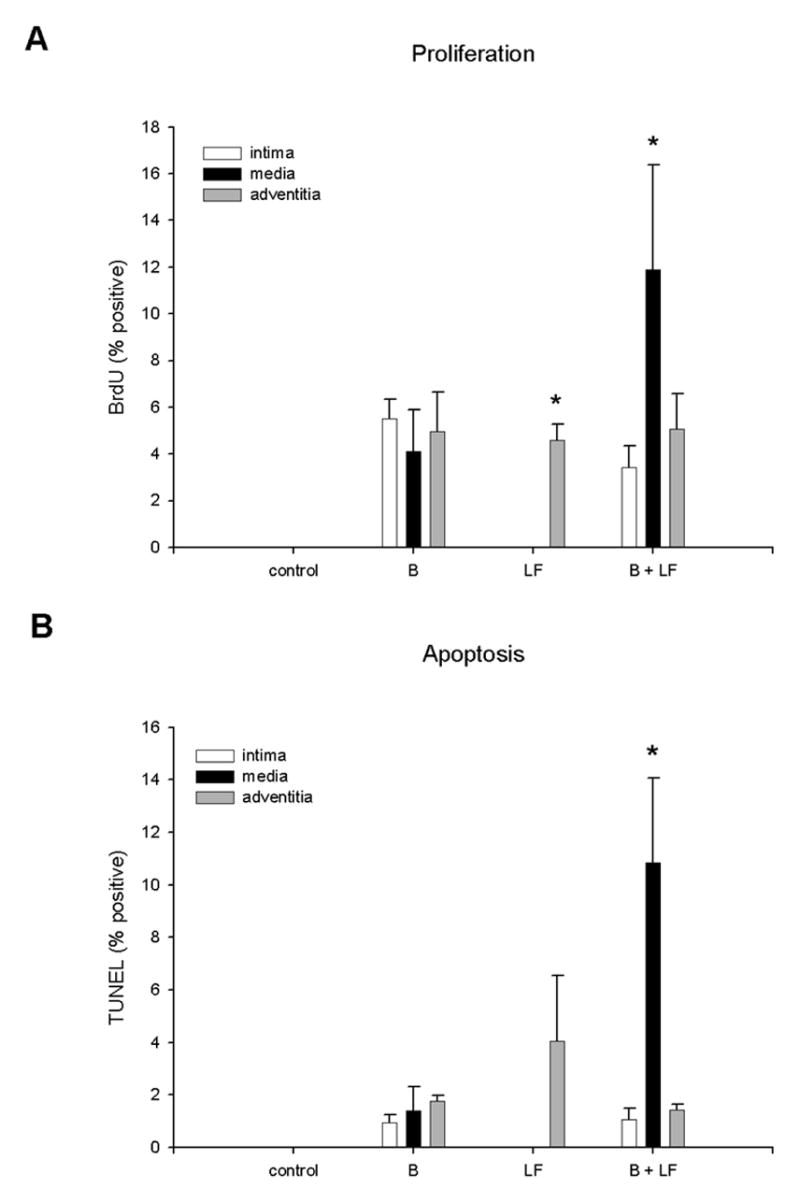
Proliferation and apoptosis in vessels (7d). A) The percentage of cells staining positively for BrdU is plotted according to location within the vessel wall. The difference between the adventitia and the other layers in the LF group is significant (*; p=.005, post-hoc); the difference between the media in the B+LF group and the media of the other groups is significant (*; p=.02, post-hoc; n=12). B) The percentage of cells staining positively for TUNEL is plotted according to location within the vessel wall. The difference between the media in the B+LF group and the media of the other groups is significant (*; p=.01, post-hoc).
3.3. Changes in medial and adventitial Nogo-B levels correlate with neointimal expansion, but not adaptive remodeling
Since reduced Nogo-B immunoreactivity correlates with carotid arterial injury and Nogo-B deficient mice have accelerated neointima formation after wire injury,(Acevedo, Yu et al. 2004) we determined the pattern of Nogo-B expression in rabbit arteries exposed to injury with and without flow changes. In control vessels, Nogo-B was strongly expressed in the intima and media with little protein in the adventitia (first panels, top and bottom in Figure 3A, with quantification in Figure 3B). Treatment with B injury reduced immunoreactive Nogo-B in the intima and media, and increased Nogo-B levels in the adventitia (second panels). Treatment with LF also reduced Nogo-B levels in the intima but did not change the levels in the media or adventitia (third panels). However, with treatment with both B+LF, Nogo-B levels were reduced in both intima and media and increased in adventitia compared with control or LF alone. These results show that balloon injury of rabbit carotid arteries, similar to wire injury of mouse carotid arteries, results in increased cellular proliferation, apoptosis and decreased Nogo-B levels in the intima and media. Low flow-stimulated inward remodeling decreased intimal Nogo-B levels but is not associated with other indices of vessel injury (proliferation, apoptosis or changes in medial Nogo-B). However, after combined balloon injury with reduced flow, the adaptative remodeling triggered by flow alone is surpassed by the response to injury and Nogo-B levels in the vessel wall are similar to B alone.
Figure 3.
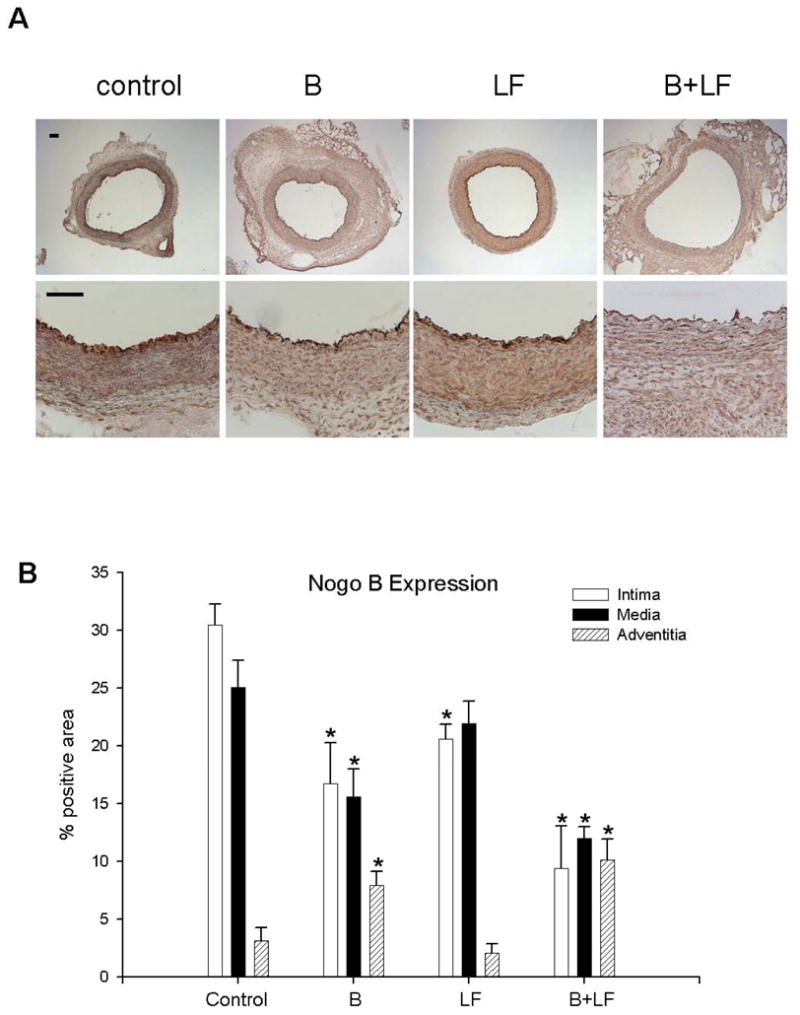
Nogo-B expression in the rabbit carotid artery (7d). A) Representative photomicrographs demonstrating immunohistochemistry for Nogo-B in the rabbit carotid artery (day 7), treated with: control, B, LF, B+LF. Scale bar, 100 μm. B) Quantitative analysis of Nogo-B density in the intima, media and adventitia.
Finally, to determine the identity of cells in the media that expressed Nogo-B, we used immunofluorescence microscopy to colocalize Nogo-B with smooth muscle alpha-actin. Immunoreactive Nogo-B (green channel) and alpha-actin (red channel) colocalized (yellow color) in the media of all vessels, consistent with the presence of Nogo-B in medial smooth muscle cells (Figure 4A, top panels). In vessels where Nogo-B levels were higher in the adventitia, i.e. with B or B+LF treatments, adventitial Nogo-B did not colocalize with alpha-actin (Figure 4A) but partially colocalized with the macrophage marker RAM-11 (Figure 4B), suggesting that at least some of the Nogo-B-containing adventitial cells in balloon injured vessels were macrophages (see Figure 4C for quantification of Nogo-B and RAM-11 positive cells in the adventitia).
Figure 4.
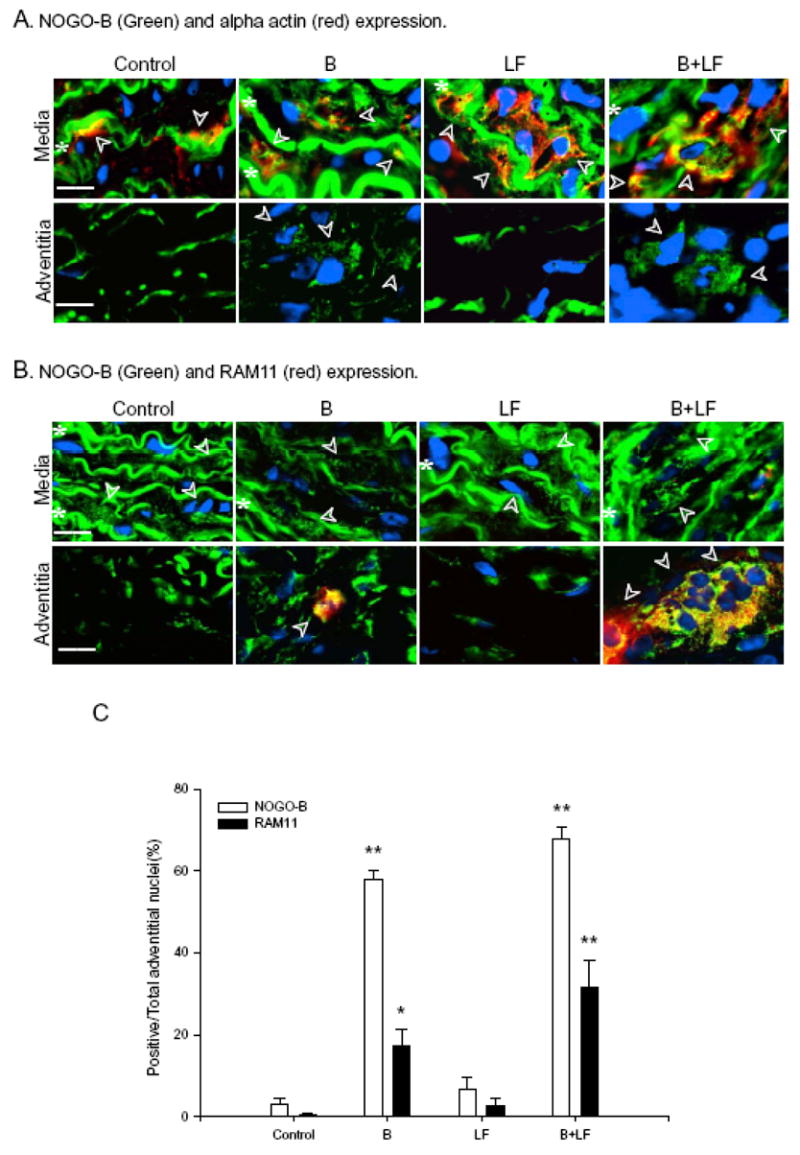
Nogo-B localization in the rabbit carotid artery (7d). A) Representative photomicrographs demonstrating immunofluorescence for Nogo-B (green), alpha-actin (red), or both merged (yellow) in the rabbit carotid artery (day 7), treated with: control, B, LF, B+LF. Nuclei are indicated by DAPI (blue) fluorescence. Row I: Media; Row II: Adventitia. Scale bar, 20 μm. Arrowheads represent cells staining for Nogo-B and alpha-actin (media), and cells staining for Nogo-B but not alpha-actin (adventitia); white stars represent autofluorescence of the medial elastic laminae. B) Representative photomicrographs demonstrating merged immunofluorescence for Nogo-B (green), RAM-11 (red), or both merged (yellow); and nuclei with DAPI (blue). Row I: Media; Row II: Adventitia. Scale bar, 20 μm. Arrowheads represent cells staining for Nogo-B but not RAM-11 (media), and cells staining for both Nogo-B and RAM-11 (adventitia); white stars represent autofluorescence of the medial elastic laminae. C) Bar graph demonstrating the percentage of cells staining positively for Nogo-B (white bars) and RAM-11 (black bars) in the adventitia of rabbits treated with: control, B, LF, B+LF.
4. Discussion
We demonstrate in the rabbit carotid artery that balloon injury-induced neointimal hyperplasia is accompanied by increased cell turnover in all layers of the vessel wall and, although Nogo-B levels are reduced in the intima and media, Nogo-B levels are enhanced in the adventitia. Low flow-induced inward remodeling in the absence of neointima stimulates adventitial cell turnover and apoptosis. This is associated with a decrease in intimal Nogo-B, but no change in medial or adventitial Nogo-B levels suggesting that low flow-induced inward remodeling differs mechanistically from vessel injury. Finally, we demonstrate that low flow significantly augments balloon injury-induced neointimal hyperplasia and is accompanied by increased medial cell turnover, reduced intimal and medial Nogo-B expression, and increased adventitial Nogo-B expression in macrophages, consistent with enhanced response to injury. These data support the idea that Nogo-B is an endogenous regulator of vascular inflammation and that the loss of Nogo-B correlates with interventions that trigger intimal remodeling or neointimal expansion.
The role of shear stress in arterial hemodynamics and response to injury has been extensively studied (Dewey, Bussolari et al. 1981; Zarins, Giddens et al. 1983; Zarins, Zatina et al. 1987; Traub and Berk 1998; Garcia-Cardena, Comander et al. 2001; Paszkowiak and Dardik 2003). The rabbit carotid artery injury model is established and has provided mechanistic insight into the role of reduced shear stress in negative remodeling (Langille and O'Donnell 1986; Langille, Bendeck et al. 1989). This model has been criticized since neointima does not form in the presence of low flow (Korshunov and Berk 2003). However, in the presence of both balloon injury and low flow, both inward remodeling and neointimal hyperplasia are augmented (Ward, Tsao et al. 2003; Hanratty, Murrell et al. 2004). We demonstrate significantly enhanced neointimal hyperplasia in the presence of low flow, compared to the response to balloon injury alone (Figure 1G) thus demonstrating the relevance of the rabbit carotid artery response to injury as a model of human stenosis that often occur in tandem fashion, with resultant low flow.
Although the contribution of the different layers of the vessel wall is apparent in this study, the role of the adventitia has traditionally been underappreciated. Apoptosis occurs during inward vessel remodeling; however, the cell layer in which it occurs is not consistently localized in some studies (Dimmeler and Zeiher 2000; Kalra and Miller 2000; Sata, Maejima et al. 2000; Sho, Sho et al. 2001). For example, Sata et al. demonstrate medial apoptosis in response to wire injury in the mouse but also present data that there appears to be extensive apoptosis in the adventitia (Sata, Maejima et al. 2000). However, many studies have demonstrated a role for the adventitia in the response to injury (Booth, Martin et al. 1989; Scott, Cipolla et al. 1996; Shi, O'Brien et al. 1996; Shi, Pieniek et al. 1996; Li, Chen et al. 2000; Zalewski, Shi et al. 2002; Hu, Zhang et al. 2004; Stenmark, Davie et al. 2006). These studies suggest that the adventitia plays an active role in the vessel’s response to injury, and may serve as a source of both cells and mediators to recruit additional cell types (Gutterman 1999; Zalewski, Shi et al. 2002; Hu, Zhang et al. 2004; Stenmark, Davie et al. 2006). Recent studies in the mouse have demonstrated changes in the adventitial volume in response to flow (Korshunov and Berk 2003). In addition, the adventitia may be an optimal site to apply therapeutic agents, compared to application within the lumen (Kalra, Jost et al. 2000). Although low flow increases macrophage infiltration in the media and adventitia in experimental aneurysms (Sho, Sho et al. 2004), macrophages were not detected in the non-aneurysmal rabbit carotid arteries in the present study. Both proliferation and apoptosis in response to low flow are maximal in the adventitia, compared to other layers of the vessel wall (Figure 2). Our results differ from previous studies in a similar model that reported intimal and medial apoptosis (Cho, Mitchell et al. 1997), possibly since we examine adult, rather than young immature, rabbits.
Nogo-B is a ubiquitous member of the reticulon protein family; Nogo-A and –C isoforms are generally restricted to the central nervous system (GrandPre, Nakamura et al. 2000). The function of Nogo is unknown, even though it may interact with diverse proteins such as Nogo-receptor, Bcl-2, Bcl-XL, myelin basic protein, and alpha-tubulin (Teng, Ling et al. 2004). Nogo-B is found on the cell surface and has been recently postulated to form a channel or transporter, although its ability to interact with MAPKAP-K2 and the p38 pathway may imply diverse intracellular roles (Dodd, Niederoest et al. 2005; Rousseau, Peggie et al. 2005). Nogo-B is found in vascular endothelial cells and smooth muscle cells, and is down regulated in vessel injury and neointimal hyperplasia (Acevedo, Yu et al. 2004). Here we confirm that Nogo-B is down regulated in the intima and media in response to balloon injury and demonstrate its medial preservation in low flow-induced inward remodeling (Figure 3). This data argue in favor of the idea that vessel remodeling in response to flow occurs via separate pathways than those stimulated in pathological responses to injury, leading to neointimal hyperplasia.
We also demonstrate increased Nogo-B levels in the adventitia of balloon-injured vessels (Figure 3). Since the cells expressing Nogo-B do not express alpha-actin, we believe that infiltrating cells, such as macrophages, may express Nogo-B in the adventitia of balloon-injured vessels. These results are consistent with our finding of increased inflammation and edema in the adventitia of these vessels (Figure 3), as well as colocalization of Nogo-B with RAM-11 in adventitial cells (Figure 4). Previous studies have demonstrated that alpha-actin containing adventitial cells – “adventitial myofibroblasts” – contribute to neointima formation (Scott, Cipolla et al. 1996; Shi, O'Brien et al. 1996; Shi, Pieniek et al. 1996); our results do not exclude these findings, but suggest an additional cell type that might contribute to vascular remodeling. Since macrophages express Nogo-B (Figure 4), these cells may be a source for adventitial Nogo-B; the ability of Nogo-B to stimulate vascular cell migration suggests a role for the adventitia in the response to vessel injury and adaptation (Acevedo, Yu et al. 2004). The inability of macrophages to enter the vessel media, restricting them to the adventitia, may be a consequence of the immunoprivileged properties of the media (Dal Canto, Swanson et al. 2001).
These results suggest that the loss of Nogo-B is a good biomarker for injury-induced neointimal hyperplasia that is distinct from low flow-induced inward remodeling. Precisely how the loss of Nogo-B promotes intimal expansion in response to injury is not known, but our data is consistent with a model in which Nogo-B serves as a negative regulator of PDGF induced smooth muscle migration (Acevedo, Yu et al. 2004). Reduced intimal and preserved medial Nogo-B levels during low flow-induced inward remodeling suggest a role for medial smooth muscle cells containing Nogo-B in the prevention of intimal expansion in the absence of injury. However, the mechanism by which endothelial cells sense low flow and have reduced Nogo-B levels during inward remodeling is not clear. Taken together, these results suggest that injury-induced neointimal hyperplasia is distinct from low flow-induced inward remodeling and that therapeutic strategies based solely on inhibition of neointimal hyperplasia may only partially account for lumen loss in complex arterial disease.
Acknowledgments
This work was supported in part by an American College of Surgeons Faculty Research Fellowship (AD), the Pacific Vascular Research Foundation EJ Wylie Award (AD), and the Peripheral Vascular Surgery Society William J. von Liebig Vascular Academic Award (AD). This material is the result of work partially supported by the National Institutes of Health Career Development Award HL 079927 / American Vascular Association William J. von Liebig Award as well as with resources and the use of facilities at the VA Connecticut Healthcare System, West Haven, CT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acevedo L, Yu J, et al. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004;10(4):382–8. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- Austin GE, Ratliff NB, et al. Intimal proliferation of smooth muscle cells as an explanation for recurrent coronary artery stenosis after percutaneous transluminal coronary angioplasty. J Am Coll Cardiol. 1985;6(2):369–75. doi: 10.1016/s0735-1097(85)80174-1. [DOI] [PubMed] [Google Scholar]
- Bauters C, Isner JM. The biology of restenosis. Prog Cardiovasc Dis. 1997;40(2):107–16. doi: 10.1016/s0033-0620(97)80003-5. [DOI] [PubMed] [Google Scholar]
- Booth RF, Martin JF, et al. Rapid development of atherosclerotic lesions in the rabbit carotid artery induced by perivascular manipulation. Atherosclerosis. 1989;76(2–3):257–68. doi: 10.1016/0021-9150(89)90109-3. [DOI] [PubMed] [Google Scholar]
- Cho A, Mitchell L, et al. Effects of changes in blood flow rate on cell death and cell proliferation in carotid arteries of immature rabbits. Circ Res. 1997;81(3):328–37. doi: 10.1161/01.res.81.3.328. [DOI] [PubMed] [Google Scholar]
- Dal Canto AJ, Swanson PE, et al. IFN-gamma action in the media of the great elastic arteries, a novel immunoprivileged site. J Clin Invest. 2001;107(2):R15–22. doi: 10.1172/JCI11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey CF, Jr, Bussolari SR, et al. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng. 1981;103(3):177–85. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Zeiher AM. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ Res. 2000;87(6):434–9. doi: 10.1161/01.res.87.6.434. [DOI] [PubMed] [Google Scholar]
- Dodd DA, Niederoest B, et al. Nogo-A, -B, and -C are found on the cell surface and interact together in many different cell types. J Biol Chem. 2005;280(13):12494–502. doi: 10.1074/jbc.M411827200. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Comander J, et al. Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc Natl Acad Sci U S A. 2001;98:4478–4485. doi: 10.1073/pnas.071052598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girerd X, London G, et al. Remodeling of the radial artery in response to a chronic increase in shear stress. Hypertension. 1996;27(3 Pt 2):799–803. doi: 10.1161/01.hyp.27.3.799. [DOI] [PubMed] [Google Scholar]
- GrandPre T, Nakamura F, et al. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403(6768):439–44. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- Gutterman DD. Adventitia-dependent influences on vascular function. Am J Physiol. 1999;277(4 Pt 2):H1265–72. doi: 10.1152/ajpheart.1999.277.4.H1265. [DOI] [PubMed] [Google Scholar]
- Guzman RJ, Abe K, et al. Flow-induced arterial enlargement is inhibited by suppression of nitric oxide synthase activity in vivo. Surgery. 1997;122(2):273–9. doi: 10.1016/s0039-6060(97)90018-0. discussion 279–80. [DOI] [PubMed] [Google Scholar]
- Hanratty CG, Murrell M, et al. Low flow promotes instent intimal hyperplasia.Comparison with lumen loss in balloon-injured and uninjured vessels and the effects of the antioxidant pyrrolidine dithiocarbamate. Atherosclerosis. 2004;177(2):269–74. doi: 10.1016/j.atherosclerosis.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Harmon KJ, Couper LL, et al. Strain-dependent vascular remodeling phenotypes in inbred mice. Am J Pathol. 2000;156(5):1741–8. doi: 10.1016/S0002-9440(10)65045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Zhang Z, et al. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J Clin Invest. 2004;113(9):1258–65. doi: 10.1172/JCI19628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuta T, Currier JW, et al. Differences in compensatory vessel enlargement, not intimal formation, account for restenosis after angioplasty in the hypercholesterolemic rabbit model. Circulation. 1994;89(6):2809–15. doi: 10.1161/01.cir.89.6.2809. [DOI] [PubMed] [Google Scholar]
- Kalra M, Jost CJ, et al. Adventitial versus intimal liposome-mediated ex vivo transfection of canine saphenous vein grafts with endothelial nitric oxide synthase gene. J Vasc Surg. 2000;32(6):1190–200. doi: 10.1067/mva.2000.109211. [DOI] [PubMed] [Google Scholar]
- Kalra M, V, Miller M. Early remodeling of saphenous vein grafts: proliferation, migration and apoptosis of adventitial and medial cells occur simultaneously with changes in graft diameter and blood flow. J Vasc Res. 2000;37(6):576–84. doi: 10.1159/000054091. [DOI] [PubMed] [Google Scholar]
- Kamiya A, Togawa T. Adaptive regulation of wall shear stress to flow change in the canine carotid artery. Am J Physiol. 1980;239(1):H14–21. doi: 10.1152/ajpheart.1980.239.1.H14. [DOI] [PubMed] [Google Scholar]
- Kohler TR, Jawien A. Flow affects development of intimal hyperplasia after arterial injury in rats. Arterioscler Thromb. 1992;12(8):963–71. doi: 10.1161/01.atv.12.8.963. [DOI] [PubMed] [Google Scholar]
- Korshunov VA, Berk BC. Flow-induced vascular remodeling in the mouse: a model for carotid intima-media thickening. Arterioscler Thromb Vasc Biol. 2003;23(12):2185–91. doi: 10.1161/01.ATV.0000103120.06092.14. [DOI] [PubMed] [Google Scholar]
- Krams R, Wentzel JJ, et al. Shear stress in atherosclerosis, and vascular remodelling. Semin Interv Cardiol. 1998;3(1):39–44. [PubMed] [Google Scholar]
- Kuntz RE, Gibson CM, et al. Generalized model of restenosis after conventional balloon angioplasty, stenting and directional atherectomy. J Am Coll Cardiol. 1993;21(1):15–25. doi: 10.1016/0735-1097(93)90712-a. [DOI] [PubMed] [Google Scholar]
- Lafont A, Guzman LA, et al. Restenosis after experimental angioplasty. Intimal, medial, and adventitial changes associated with constrictive remodeling. Circ Res. 1995;76(6):996–1002. doi: 10.1161/01.res.76.6.996. [DOI] [PubMed] [Google Scholar]
- Langille BL, Bendeck MP, et al. Adaptations of carotid arteries of young and mature rabbits to reduced carotid blood flow. Am J Physiol. 1989;256(4 Pt 2):H931–9. doi: 10.1152/ajpheart.1989.256.4.H931. [DOI] [PubMed] [Google Scholar]
- Langille BL, O'Donnell F. Reductions in arterial diameter produced by chronic decreases in blood flow are endothelium-dependent. Science. 1986;231(4736):405–7. doi: 10.1126/science.3941904. [DOI] [PubMed] [Google Scholar]
- Li G, Chen SJ, et al. Direct in vivo evidence demonstrating neointimal migration of adventitial fibroblasts after balloon injury of rat carotid arteries. Circulation. 2000;101(12):1362–5. doi: 10.1161/01.cir.101.12.1362. [DOI] [PubMed] [Google Scholar]
- Ohashi N, Matsumori A, et al. Role of p38 mitogen-activated protein kinase in neointimal hyperplasia after vascular injury. Arterioscler Thromb Vasc Biol. 2000;20(12):2521–6. doi: 10.1161/01.atv.20.12.2521. [DOI] [PubMed] [Google Scholar]
- Paszkowiak JJ, Dardik A. Arterial wall shear stress: observations from the bench to the bedside. Vasc Endovascular Surg. 2003;37(1):47–57. doi: 10.1177/153857440303700107. [DOI] [PubMed] [Google Scholar]
- Post MJ, Borst C, et al. The relative importance of arterial remodeling compared with intimal hyperplasia in lumen renarrowing after balloon angioplasty. A study in the normal rabbit and the hypercholesterolemic Yucatan micropig. Circulation. 1994;89(6):2816–21. doi: 10.1161/01.cir.89.6.2816. [DOI] [PubMed] [Google Scholar]
- Post MJ, Borst C, et al. Arterial remodeling in atherosclerosis and restenosis: a vague concept of a distinct phenomenon. Atherosclerosis. 1995;118(Suppl):S115–23. [PubMed] [Google Scholar]
- Post MJ, de Smet BJ, et al. Arterial remodeling after balloon angioplasty or stenting in an atherosclerotic experimental model. Circulation. 1997;96(3):996–1003. doi: 10.1161/01.cir.96.3.996. [DOI] [PubMed] [Google Scholar]
- Qin F, Dardik H, et al. Remodeling and suppression of intimal hyperplasia of vascular grafts with a distal arteriovenous fistula in a rat model. J Vasc Surg. 2001;34(4):701–6. doi: 10.1067/mva.2001.116804. [DOI] [PubMed] [Google Scholar]
- Rectenwald JE, Moldawer LL, et al. Direct evidence for cytokine involvement in neointimal hyperplasia. Circulation. 2000;102(14):1697–702. doi: 10.1161/01.cir.102.14.1697. [DOI] [PubMed] [Google Scholar]
- Rousseau S, Peggie M, et al. Nogo-B is a new physiological substrate for MAPKAP-K2. Biochem J. 2005;391(Pt 2):433–40. doi: 10.1042/BJ20050935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sata M, Maejima Y, et al. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32(11):2097–104. doi: 10.1006/jmcc.2000.1238. [DOI] [PubMed] [Google Scholar]
- Schwartz RS. Pathophysiology of restenosis: interaction of thrombosis, hyperplasia, and/or remodeling. Am J Cardiol. 1998;81(7A):14E–17E. doi: 10.1016/s0002-9149(98)00191-x. [DOI] [PubMed] [Google Scholar]
- Scott NA, Cipolla GD, et al. Identification of a potential role for the adventitia in vascular lesion formation after balloon overstretch injury of porcine coronary arteries. Circulation. 1996;93(12):2178–87. doi: 10.1161/01.cir.93.12.2178. [DOI] [PubMed] [Google Scholar]
- Shi Y, O'Brien JE, et al. Adventitial myofibroblasts contribute to neointimal formation in injured porcine coronary arteries. Circulation. 1996;94(7):1655–64. doi: 10.1161/01.cir.94.7.1655. [DOI] [PubMed] [Google Scholar]
- Shi Y, Pieniek M, et al. Adventitial remodeling after coronary arterial injury. Circulation. 1996;93(2):340–8. doi: 10.1161/01.cir.93.2.340. [DOI] [PubMed] [Google Scholar]
- Sho E, Sho M, et al. Hemodynamic forces regulate mural macrophage infiltration in experimental aortic aneurysms. Exp Mol Pathol. 2004;76(2):108–16. doi: 10.1016/j.yexmp.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Sho E, Sho M, et al. Blood flow decrease induces apoptosis of endothelial cells in previously dilated arteries resulting from chronic high blood flow. Arterioscler Thromb Vasc Biol. 2001;21(7):1139–45. doi: 10.1161/hq0701.092118. [DOI] [PubMed] [Google Scholar]
- Song RH, Kocharyan HK, et al. Increased flow and shear stress enhance in vivo transforming growth factor-beta1 after experimental arterial injury. Arterioscler Thromb Vasc Biol. 2000;20(4):923–30. doi: 10.1161/01.atv.20.4.923. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Davie N, et al. Role of the adventitia in pulmonary vascular remodeling. Physiology Bethesda. 2006;(21):134–45. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
- Teng FY, Ling BM, et al. Inter- and intracellular interactions of Nogo: new findings and hypothesis. J Neurochem. 2004;89(4):801–6. doi: 10.1111/j.1471-4159.2004.02366.x. [DOI] [PubMed] [Google Scholar]
- Traub O, Berk BC. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler Thromb Vasc Biol. 1998;18(5):677–85. doi: 10.1161/01.atv.18.5.677. [DOI] [PubMed] [Google Scholar]
- Tronc F, Wassef M, et al. Role of NO in flow-induced remodeling of the rabbit common carotid artery. Arterioscler Thromb Vasc Biol. 1996;16(10):1256–62. doi: 10.1161/01.atv.16.10.1256. [DOI] [PubMed] [Google Scholar]
- Tuttle JL, Nachreiner RD, et al. Shear level influences resistance artery remodeling: wall dimensions, cell density, and eNOS expression. Am J Physiol Heart Circ Physiol. 2001;281(3):H1380–9. doi: 10.1152/ajpheart.2001.281.3.H1380. [DOI] [PubMed] [Google Scholar]
- Ward MR, Tsao PS, et al. Low blood flow after angioplasty augments mechanisms of restenosis: inward vessel remodeling, cell migration, and activity of genes regulating migration. Arterioscler Thromb Vasc Biol. 2001;21(2):208–13. doi: 10.1161/01.atv.21.2.208. [DOI] [PubMed] [Google Scholar]
- Ward MR, Tsao PS, et al. Flow-responsive remodeling after angioplasty is enhanced by high cholesterol diet. Prevention with pyrrolidine dithiocarbamate. Atherosclerosis. 2003;168(2):333–41. doi: 10.1016/s0021-9150(03)00103-5. [DOI] [PubMed] [Google Scholar]
- Wentzel JJ, Gijsen FJ, et al. Shear stress, vascular remodeling and neointimal formation. J Biomech. 2003;36(5):681–8. doi: 10.1016/s0021-9290(02)00446-3. [DOI] [PubMed] [Google Scholar]
- Zalewski A, Shi Y, et al. Diverse origin of intimal cells: smooth muscle cells, myofibroblasts, fibroblasts, and beyond?". Circ Res. 2002;91(8):652–5. doi: 10.1161/01.res.0000038996.97287.9a. [DOI] [PubMed] [Google Scholar]
- Zarins C, Giddens D, et al. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res. 1983;53:502–514. doi: 10.1161/01.res.53.4.502. [DOI] [PubMed] [Google Scholar]
- Zarins CK, Zatina MA, et al. Shear stress regulation of artery lumen diameter in experimental atherogenesis. J Vasc Surg. 1987;5(3):413–20. [PubMed] [Google Scholar]