

Abstract
Metastatic renal cell carcinoma (RCC) resulting from the hereditary loss of the von Hippel Lindau (VHL) tumor suppressor gene is the leading cause of death in VHL patients due to the deleterious effects of the metastatic tumor(s). VHL functions in the destruction of the alpha subunits of the heterodimeric transcription factor, Hypoxia-Inducible Factor (HIF-1α and HIF-2α), in normoxic conditions. When VHL function is lost, HIF-α protein is stabilized, and target hypoxia-inducible genes are transcribed. The process of tumor invasion and metastasis involves the destruction of the extracellular matrix (ECM), which is accomplished primarily by the matrix metalloproteinase (MMP) family of enzymes. Here, we describe a connection between the loss of VHL tumor suppressor function and the upregulation of Membrane Type-1 MMP (MT1-MMP) gene expression and protein. Specifically, MT1-MMP is upregulated in VHL−/− RCC cells through an increase in gene transcription, which is mediated by the cooperative effects of the transcription factors, HIF-2 and Sp1. Further, we identify a functional HIF binding site (HBS) in the proximal promoter of MT1-MMP. To our knowledge, this is the first report to show direct regulation of MT1-MMP by HIF-2 and to provide a direct link between the loss of VHL tumor suppressor function and an increase in MMP gene and protein expression.
Keywords: von Hippel Lindau, MT1-MMP, HIF-2α, renal cell carcinoma
Introduction
Von Hippel-Lindau (VHL) disease resulting from the loss of the VHL tumor suppressor gene is a dominantly inherited familial cancer affecting several organ systems (Hes et al., 2001). Life expectancy is greatly reduced for almost 45% of all VHL patients who will develop metastatic renal cell carcinoma (RCC) in addition to other VHL primary tumors (Linehan et al., 2003). As such, metastatic RCC is the leading cause of death in VHL patients due to complications associated with the metastatic tumors (Maher & Kaelin, 1997; Richards et al., 1998; Singh et al., 2001). Interestingly, almost 80% of sporadic renal cell carcinoma is the result of mutations in the VHL gene (Barry & Krek, 2004).
VHL mediates the cell’s response to changes in oxygen tension by regulating the protein stability of Hypoxia Inducible Factor α (HIF-1α and HIF-2α also known as EPAS1). In normoxia, VHL binds HIF-α through the recognition of two hydroxy-proline residues and polyubiquitinates HIF-α to target it for proteosomal degradation (Barry & Krek, 2004; Kaelin, 2002). When a cell enters hypoxia, HIF-α protein is stabilized and partners with HIF-β (a constitutively expressed protein also known as ARNT1) to form the functional transcription factor HIF, which activates hypoxia-inducible genes such as those involved in angiogenesis and glycolysis (i.e., VEGF, EPO1, PDGF, GLUT1) (Harris, 2002). In VHL disease, the function of VHL is lost; therefore, the cell accumulates HIF-α protein and downstream target genes are thus transcribed.
To achieve metastasis, a tumor cell must disrupt cell-cell and cell-ECM (extracellular matrix) contacts, migrate through stromal tissue, invade the basement membrane, and enter/exit the blood stream (Bogenrieder & Herlyn, 2003). These steps are accomplished primarily by the activity of the matrix metalloproteinases (MMPs) family of enzymes (Egeblad & Werb, 2002; Seiki, 2003). Elevated levels of MMPs have been correlated with the invasiveness of several human cancers, as well as with other tumor phenotypes such as angiogenesis, cell proliferation, and resistance to apoptosis (Egeblad & Werb, 2002; McCawley & Matrisian, 2001). Most MMPs are secreted from cells as zymogens requiring extracellular activation by serine proteases or other MMPs. In addition to proteolytic activation, normal MMP activity is controlled by tissue inhibitors of metalloproteinases (TIMPs), proteins which function by active site inhibition (Visse & Nagase, 2003).
Membrane Type-1 MMP (MT1-MMP), a membrane bound MMP, degrades several ECM components, controls cell-ECM contacts, localizes to the leading edge of invasive cells, as well as activates latent MMP-2 (Hotary et al., 2000; Lehti et al., 1998; Mori et al., 2002; Seiki, 2003). Interestingly, TIMP-2 plays a role in the mechanism of pro-MMP-2 activation by MT1-MMP, which requires a complex of all three proteins each at a specific stiochiometric ratio (Strongin et al., 1995). Increased expression of MT1-MMP has been measured in several human carcinomas, and its expression correlates with increased invasiveness and growth of tumor cells (Hotary et al., 2003; Iida et al., 2004; Kitagawa et al., 1999; Sato et al., 1994). Importantly, MT1-MMP expression is significantly higher in RCC tumors of later pathologic stage compared to early stage (Kitagawa et al., 1999).
Although VHL has been implicated in playing a role in RCC tumor invasion and metastasis, more studies are required to clearly define the mechanisms involved (Barry & Krek, 2004). Koochekpour et al. described an increase in basal MMP-2 and MMP-9 expression as well as a decrease in TIMP levels in RCC cells lacking VHL expression (Koochekpour et al., 1999). They further demonstrated that MMP activity drives the invasive capability of VHL renal cell carcinoma cells in an in vitro system. Because MT1-MMP activates pro-MMP-2, we hypothesized that MT1-MMP expression is also regulated by VHL in RCC cells. To investigate mechanisms of MT1-MMP regulation in VHL renal cell carcinoma, we used cell lines derived from the human 786-0 renal cell carcinoma cell line.
Here we report that in the absence of VHL, RCC cells overexpress MT1-MMP on the levels of gene and protein expression. Our data support a model in which the loss of the VHL tumor suppressor and subsequent stability of HIF-2α leads to induction of MT1-MMP expression. We define the mechanism of upregulation as direct transcriptional activation of MT1-MMP by HIF-2 in coordination with Sp1. To our knowledge, this is the first report to demonstrate MT1-MMP as a HIF regulated gene and to suggest a role for MT1-MMP in VHL RCC tumor invasion and metastasis.
Results
MT1-MMP is overexpressed in RCC cells lacking VHL on the levels of gene and protein expression
We first analyzed MMP gene expression in RCC cells that express either wild-type VHL (WT8) or are null for VHL (pRc-9) using northern blot analysis and real-time RT-PCR. The basal and inducible (by growth factors and cytokines) expression levels of MMP-1, MMP-2, MMP-3, MMP-9, MMP-13, and MT1-MMP were measured. Basal levels of MMP-1, MMP-3, and MMP-13 were too low to detect, and although MMP-1 was inducible, no significant difference in expression was observed between cell lines (data not shown). These data suggest a lack of regulation of these MMPs by VHL. In contrast, data for expression of MMP-2 and MMP-9 agreed with those of Koochekpour et al. in that the pRc-9 cells constitutively overexpressed these MMPs compared to the WT8s (data not shown and (Koochekpour et al., 1999)). Further, we found that MT1-MMP, which has a functional relationship with MMP-2 (Seiki, 2003), was overexpressed at least 2–3 fold in the pRc-9 cell line at both the level of mRNA (Figure 1a, 1b) and protein (Figure 1c). Since MT1-MMP protein levels correlate with mRNA expression levels, we focused our studies on VHL regulation of MT1-MMP gene expression.
Figure 1.
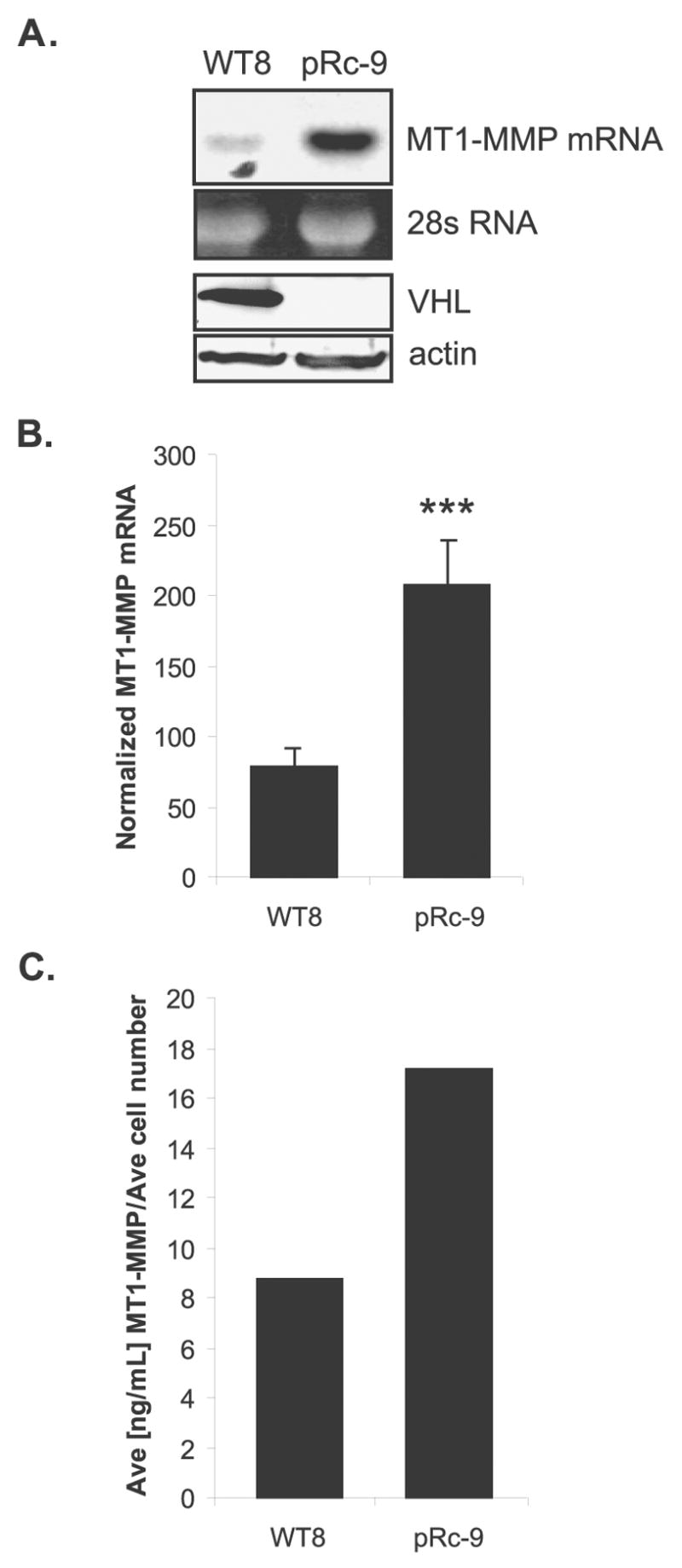
MT1-MMP overexpression in RCC cells lacking VHL. Total RNA and protein were isolated from WT8 and pRc-9 cells. (A) Northern blot analysis of MT1-MMP mRNA expression and western blot analysis of VHL protein expression in RCC cell lines. RNA levels were normalized to 28s RNA and protein levels were normalized to actin. (B) MT1-MMP mRNA expression quantitated by real-time RT-PCR normalized to GAPDH. Values represent the average of pg of MT1-MMP divided by ng of GAPDH from ≥3 individual experiments (mean +/− S.D.) ; p=0.0007. (C) MT1-MMP protein expression as quantitated by a MT1-MMP ELISA activity assay. Values represent the average of 3 samples normalized to average cell number at time of harvest. Statistical analyses were performed using the student’s t-test.
VHL expression inhibits MT1-MMP transcription and HIF-2α protein levels
To study VHL regulation of MT1-MMP transcription, we measured MT1-MMP nascent heteronuclear (hn) RNA production in the RCC cell lines using quantitative RT-PCR. The pRc-9 cells had elevated levels of MT1-MMP hnRNA compared to the WT8s (Figure 2a), demonstrating that the overexpression of MT1-MMP in the pRc-9s (see Figure 1) is a result of increased transcription in these cells. Furthermore, treatment with Actinomycin D showed no significant difference in the mRNA decay curves between the cell lines (data not shown), supporting the conclusion that the difference in MT1-MMP expression is due to transcriptional effects rather than mRNA stability effects. Next, VHL regulation of MT1-MMP gene expression was confirmed by transient overexpression of a VHL expression construct in the pRc-9 cells. Transfection efficiency was high enough to measure the effects of VHL expression on endogenous gene expression. Overexpression of wild-type VHL in the pRc-9 cells showed a dose-dependent decrease in endogenous MT1-MMP gene expression and HIF-2α protein levels, suggesting a role for HIF2α in MT1-MMP regulation (Figure 2b, 2c). To define the region of the MT1-MMP promoter that was responsive to negative regulation by VHL, a VHL expression construct was co-transfected with a 0.4kb fragment of the MT1-MMP promoter (0.4MT-luc) into HEK293 cells used for their ease of transfection. Overexpression of VHL significantly repressed expression of the MT1-MMP promoter construct, indicating that at least some elements by which VHL regulates MT1-MMP gene expression are present in the proximal promoter (Figure 2d). Similar results were obtained using the pRc-9 cells (data not shown).
Figure 2.
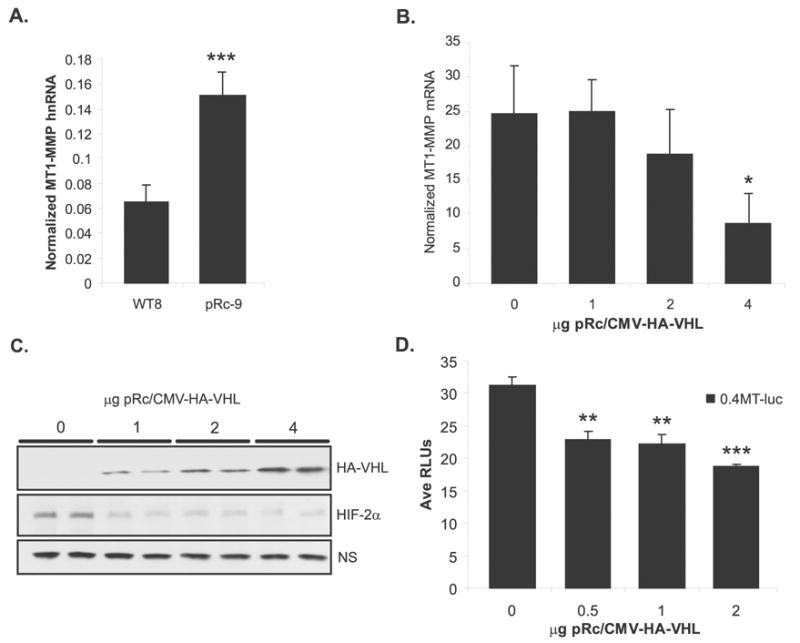
VHL down-regulates MT1-MMP and HIF-2α. (A) MT1-MMP transcription as measured by real-time RT-PCR quantization of nascent heteronuclear transcript normalized to GAPDH. Values represent the average of pg of MT1-MMP divided by ng of GAPDH from ≥3 individual experiments (mean+/− S.D.); p=0.0003. (B) MT1-MMP mRNA quantitated by real-time RT-PCR in pRc-9 cells transiently transfected with pCMV-HA-VHL. Transfection efficiency was 30–40% as determined by GFP expression. Values represent the average of pg of MT1-MMP normalized to ng of GAPDH from 3 independent experiments (mean +/− S.D.); p=0.032. (C) Lysates from the pRC-9 transfectants were subjected to immunoblotting for VHL and HIF-2α. A non-specific (NS) band present in the HIF-2α blot was used as a loading control. (D) Representative experiment from HEK 293 cells co-transfected with 0.4kb MT1-MMP promoter construct and pCMV-HA-VHL. Values represent average relative luciferase units (mean +/− S.D.); p=0.0014 (**), p=0.0001 (***). Statistical analyses were performed using the student’s t-test.
MT1-MMP transcription is regulated by HIF-2α
When VHL function is lost, HIF-α protein is stabilized, forms a heterodimer with HIF-β, and target hypoxia-inducible genes are transcribed. The RCC cell lines used in these studies express only the HIF-2α isoform (Maxwell et al., 1999), and we postulated that HIF-2α may mediate the increase in MT1-MMP transcription in the pRc-9 cells.
To test whether transient HIF-2α overexpression could activate a promoter containing HIF binding sites in a wild-type VHL background, the WT8s were co-transfected with the pTK-RE3-luc reporter with either expression constructs for pCMV-HIF2α or pCMV-HIFβ, or with the two expression constructs together. Transient overexpression of pCMV-HIF2α alone was sufficient to induce the pTK-RE3-luc reporter construct in the WT8 cells (Figure 3a). Additionally, co-expression of pCMV-HIF2α with pCMV-HIFβ did not induce the promoter further than pCMV-HIF2α alone, suggesting that endogenous HIFβ levels are sufficient to dimerize with transiently expressed HIF-2α and transactivate the reporter construct. This result is in agreement with the findings of Tian et al. (Tian et al., 1997). As expected, the expression of pCMV-HIFβ alone did not drive expression of the reporter since HIF binds DNA as a heterodimer (Jiang et al., 1996).
Figure 3.
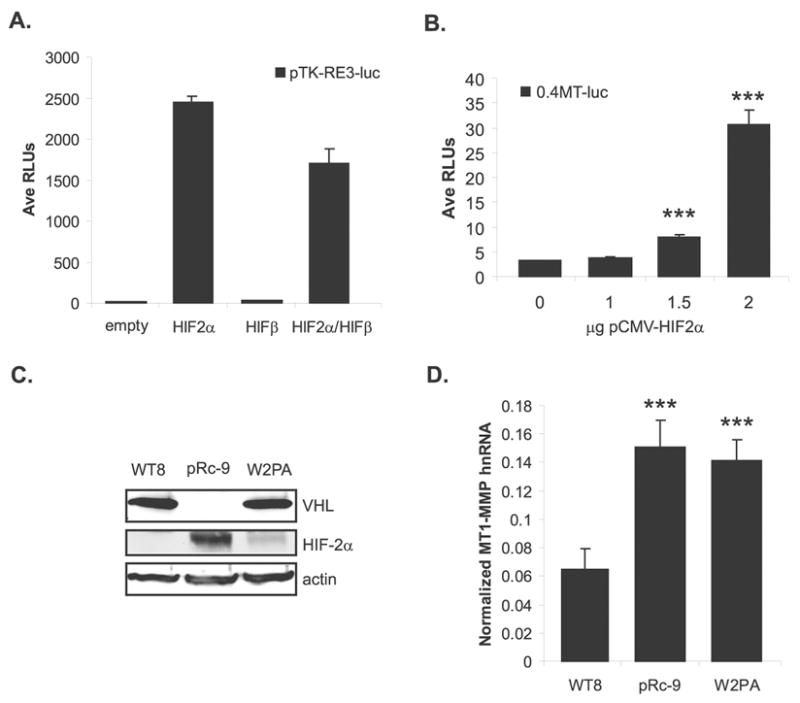
MT1-MMP transcription is regulated by HIF-2α. (A) WT8 cells co-transfected with pTK-RE3-luc and either pCMV-HIF2α, pCMV-ARNT1/HIFβ, or pCMV-HIF2α + pCMV-ARNT1/HIFβ. Total DNA was kept constant with the addition of pRc/CMV empty vector. Data are of a representative experiment and are presented as average relative luciferase units. (B) Representative experiment of WT8 cells co-transfected with 0.4MT-luc promoter construct and a dose responsive addition of pCMV-HIF2α (mean +/− S.D.). Total DNA was kept constant with the addition of pRc/CMV empty vector; p=0.0001. (C) VHL and HIF-2α protein expression in WT8, pRC-9 and W+2PA cell lines normalized to actin. (D) MT1-MMP hnRNA levels in the three RCC cell lines quantitated by real-time RT-PCR. Values represent the average of pg MT1-MMP normalized to ng GAPDH from ≥3 individual experiments (mean +/− S.D.); p<0.0001. Statistical analyses were performed using the student’s t-test.
Given these results, HIF-2α regulation of MT1-MMP in the WT8 cells was tested by transient overexpression of pCMV-HIF2α with the 0.4MT-luc promoter construct. Overexpression of HIF-2α induced the MT1-MMP promoter in the WT8 cells in a dose responsive manner (Figure 3b). Thus, forced HIF-2α expression leads to the upregulation of MT1-MMP transcription in the presence of wild-type VHL, suggesting that HIF-2 mediates MT1-MMP overexpression in VHL RCC. In order to confirm HIF-2α regulation of endogenous MT1-MMP gene expression, a third RCC cell line (W+2PA) was used that stably expressed wild-type VHL and a variant of HIF-2α, which is not degraded by VHL (Figure 3c) (Kondo et al., 2002a). Similar to the pRc-9 cells, the W+2PA cells express elevated levels of MT1-MMP hnRNA compared to the WT8 cells (Figure 3d), showing that cells, which have stable HIF-2α protein expression, overexpress MT1-MMP.
Identification of a HIF-2 binding site at -125 in the MT1-MMP promoter
To determine whether HIF-2α regulates MT1-MMP transcription directly on the MT1-MMP promoter or indirectly through the activation of another transcription factor(s) which could then target MT1-MMP, we searched the proximal 0.4kb promoter for HIF binding sites (HBSs). Two putative HBSs (HBS1−125 and HBS2+144) were identified, and the sequences are conserved in the mouse MT1-MMP promoter, suggesting possible functionality (Figure 4). The WT8 cells were co-transfected with the pCMV-HIF2α expression construct and either the 0.4kb MT1-MMP promoter construct, which contains the HBSs at −125 and +144, or a 0.1kb MT1-MMP promoter construct, which has only the HBS site at +144. Interestingly, HIF-2α was unable to induce the 0.1kb promoter fragment lending support to the HBS at −125 as the functional HIF site (Figure 5a).
Figure 4.
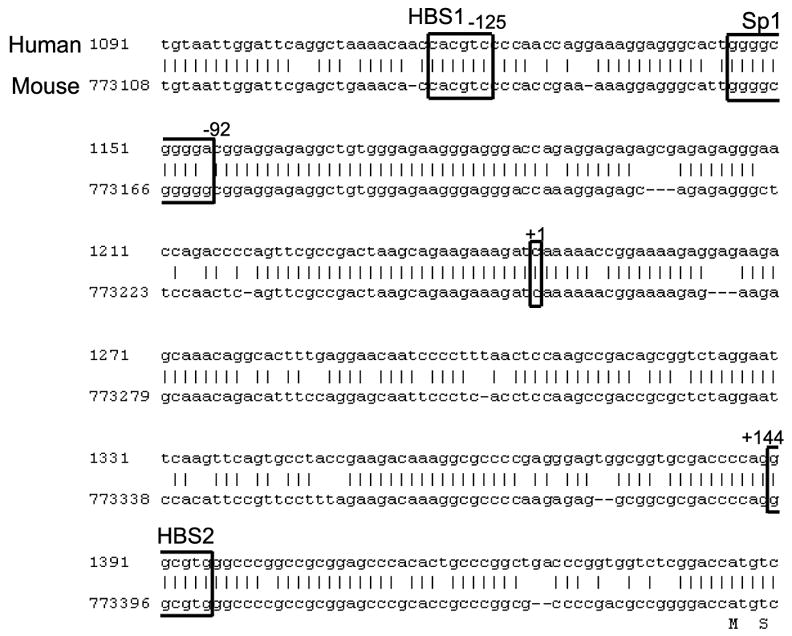
Alignment of the human (GenBank accession #AF158733) and mouse (GenBank accession #NT_039606) MT1-MMP promoters. MT1-MMP has a TATA-less promoter but contains a functional Sp1 site responsible for basal transcription at −92. Two putative HIF binding sites (HBSs) exist at −125 and +144. The HBS consensus sequence is 5’-RCGTG-3’ and both putative HBSs are conserved in the mouse. Note the −125 putative HBS is in the reverse orientation.
Figure 5.
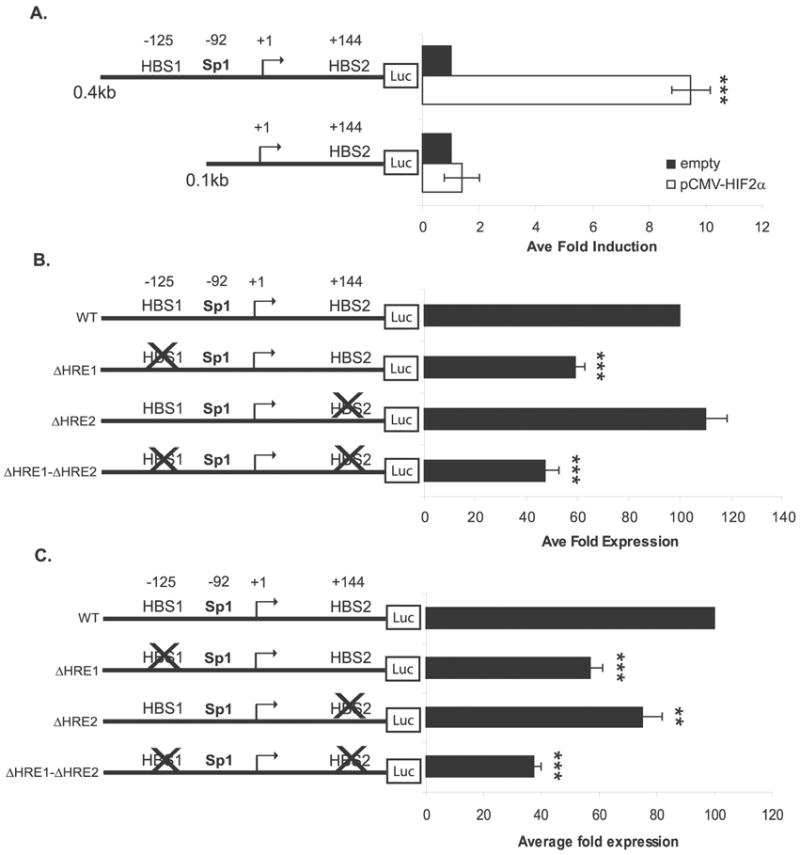
Identification of a HIF-2 binding site at −125 in the MT1-MMP promoter. (A) WT8 cells co-transfected with reporter constructs containing 0.4kb and 0.1kb of the MT1-MMP promoter and pCMV-HIF2α. Total DNA was balanced with the addition of the pRc/CMV empty vector. Data represent the average of 3 individual experiments (mean +/− S.D.); p<0.0001 (***). (B) MT1-MMP HBS mutagenesis promoter constructs were transiently co-transfected with pCMV-HIF2α in the WT8 cells. Data represent the average fold expression from three independent experiments (mean +/− S.D.); p≤0.0001 (***). (C) MT1-MMP HBS mutagenesis promoter constructs transiently expressed alone in the pRc-9 cells. Data represent the average fold expression from three independent experiments (mean +/− S.D.); p<0.0001 (***), p=0.0032 (**). Statistical analyses were performed using the student’s t-test.
To confirm this finding, multi site-directed mutagenesis was used to mutate each HBS in the MT1-MMP promoter (Table 1). Wild-type and mutated promoter constructs were transfected alone in the WT8 cells, which do not express HIF-2α protein (see Figure 3c), and no difference in expression was measured (data not shown). However, when the mutated promoter constructs were transiently co-expressed with the pCMV-HIF2α expression construct in the WT8 cells, HIF-2α induced expression was impaired by mutation of HBS1 (Figure 5b). Mutation of HBS1 (−125) reduced expression of the MT1-MMP promoter by approximately 40%, whereas mutation of HBS2 (+144) had no effect. Likewise, mutation of both HBSs did not significantly reduce expression beyond that which was observed with HBS1 mutated alone (~50% reduction), again suggesting that HBS1 is the functional HIF-2 binding site.
Table 1.
Site-directed mutagenesis of MT1-MMP promoter
| (−125) | (−92) | (−144) | |
|---|---|---|---|
| HBS1
|
Sp1
|
HBS2
|
|
| WT | 5′___CACGTC____ | GGGGCCCCCA | ____GGCGTG___3′ |
| ΔHRE1 | 5′___CTTTTC____ | GGGGCCCCCA | ____GGCGTG___3′ |
| ΔHRE2 | 5′___CACGTC____ | GGGGCCCCCA | ____GGAAAG___3′ |
| ΔHRE1 − ΔHRE2 | 5′___CTTTTC____ | GGGGCCCCCA | ____GGAAAG___3′ |
Similar results were obtained when the mutated promoter constructs were expressed alone in the pRc-9 cells (Figure 5c), which express high levels of endogenous HIF-2α (see Figure 3c), demonstrating that endogenous levels of HIF-2α resulting from the loss of VHL can regulate expression of the HBS constructs. These data give further support to HBS1 as the site by which VHL regulates MT1-MMP via a HIF-2α dependent mechanism. Interestingly, a slight but statistically significant reduction (20%) in expression of the ΔHBS2 construct was observed in the pRc-9 cells, and mutation of both HBSs reduced expression in an additive manner (40% for HBS1, 20% for HBS2, 60% for both) (Figure 5c). These data suggest that HBS2 is also regulated by HIF-2 in these cells and that this site may be functional in the pRc-9 cells. Since this effect was not observed in the WT8 cells, it is possible that overexpression of HIF-2α in the WT8s overwhelms the promoter such that minor effects on transcription, which are otherwise apparent when looking at the effects of the endogenous transcription factor (as in the pRc-9s), are missed.
Proximal Sp1 site is necessary for basal expression and full HIF-2α induction of MT1-MMP
As shown in Figure 5, mutation of HBS1 did not fully abrogate expression of MT1-MMP, suggesting that other mechanisms or factors contribute to its regulation. MT1-MMP has a TATA-less promoter, and basal expression is driven by a proximal Sp1 site at −92 from the transcriptional start site (see Figure 4 and (Lohi et al., 2000)). We confirmed that basal transcription of MT1-MMP is dependent on this Sp1 site in the WT8 cell line by measuring the expression of the 0.4MT-luc promoter construct and the 0.4MT-luc promoter construct with the Sp1 site mutated (0.4ΔSp1MT-luc) in the WT8 cells (Figure 6a). Many genes have recently been identified in which HIF-1 and Sp1 have a coordinate relationship in their transcriptional regulation, such as RORα, Endoglin, and EPO1 (Miki et al., 2004; Sanchez-Elsner et al., 2002; Sanchez-Elsner et al., 2004). Therefore, we hypothesized that HIF-2 induces MT1-MMP in coordination with Sp1. To test possible cooperativity between the Sp1 and HBS1 sites, the 0.4MT-luc and 0.4ΔSp1MT-luc constructs were co-transfected with or without pCMV-HIF-2α in the WT8 cells. Interestingly, mutation of the Sp1 site abrogated HIF-2α induction of the MT1-MMP promoter by more than 50%, suggesting that these transcription factors function coordinately in the induction of MT1-MMP (Figure 6b).
Figure 6.
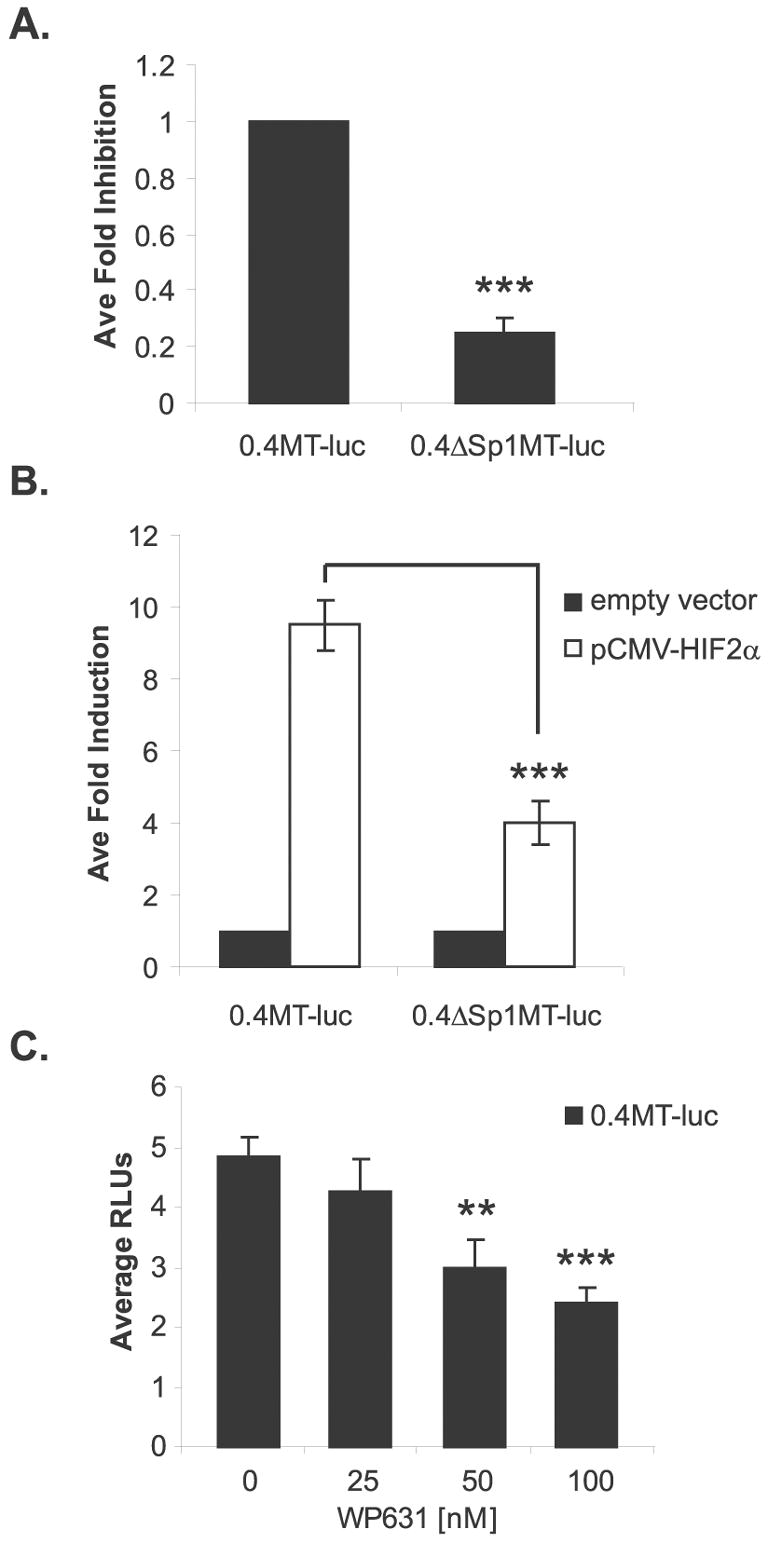
Sp1 is necessary for full HIF-2α induction of MT1-MMP. (A) WT8 cells transiently transfected with 0.4MT-luc and 0.4ΔSp1MT-luc promoter constructs. Data represent the average fold inhibition of 5 or more independent experiments (mean +/− S.D.); p<0.0001. (B) WT8 cells co-transfected with pCMV-HIF2α and either 0.4MT-luc or 0.4ΔSp1MT-luc. Total DNA was balanced with the addition of the pRc/CMV empty vector. Data represent the average fold induction of 3 individual experiments (mean +/− S.D.); p=0.0005. (C) Dose responsive treatment with Sp1 site inhibitor, WP631, of WT8 cells transiently co-transfected with 0.4MT-luc and pCMV-HIF-2α (mean S.D.); p=0.0011 (**), p<0.0001 (***). Statistical analyses were performed using the student’s t-test.
To confirm these results, we used WP631, a bisanthracycline that competitively binds to G/C rich sequence of Sp1 sites with an affinity similar to protein, to inhibit Sp1 mediated transcription at nanomolar concentrations (Gaidarova & Jimenez, 2002; Martin et al., 1999). WT8 cells were co-transfected with 0.4kb of the MT1-MMP promoter and the pCMV-HIF2α expression construct and treated with WP631. HIF-2α-induced expression of the MT1-MMP promoter was inhibited in a dose responsive manner with WP631 treatment (Figure 6c), again suggesting these transcription factors are functioning coordinately at the MT1-MMP promoter. Taken together, these results show that Sp1 is necessary for full HIF-2 induction of MT1-MMP.
Discussion
Although VHL disease manifests itself as several different types of cancer, the most common cause of morbidity and mortality in these patients is metastatic renal cell carcinoma (Richards et al., 1998; Singh et al., 2001). Less than 20% of patients with metastatic RCC will survive five years beyond diagnosis (Linehan et al., 2003). Interestingly, over 90% of RCC patients with Stage I disease will survive this five year period (Linehan et al., 2003); therefore, determining the events leading to metastatic RCC will provide therapeutic approaches to prevent progression to later stages of disease. Here we describe that, in addition to MMP-2 and -9, the loss of VHL leads to the upregulation of MT1-MMP, a membrane-tethered MMP which orchestrates many key aspects of tumor cell invasion and metastasis (Hotary et al., 2000; Mori et al., 2002; Sato et al., 1994; Seiki, 2003). Importantly, we demonstrate that the increase in MT1-MMP expression is the result of transcriptional activation by HIF-2, thereby providing a link between VHL tumor suppressor function and MT1-MMP expression.
We identified MT1-MMP as a new target of HIF-2. Thus, our data imply that MT1-MMP is a hypoxia-inducible gene and that HIF-2 regulation of MT1-MMP may play a role in other cancers in which HIF is upregulated. Since HIF-1 and HIF-2 bind to the same recognition sequence (Tian et al., 1997), it is probable that HIF-1 may induce MT1-MMP expression as well. Given the essential role of HIF in a tumor’s response to hypoxia (Harris, 2002) and the contribution of MT1-MMP to the angiogenic process and tumor cell invasion (Egeblad & Werb, 2002; Seiki, 2003), it is reasonable to hypothesize that the upregulation of MT1-MMP documented in human cancers occurs in response to the hypoxic environment within the growing tumor. In fact, hypoxia has been shown to increase MT1-MMP expression in other cell types by currently undefined mechanisms (Annabi et al., 2003; Kondo et al., 2002b; Ottino et al., 2004).
However, it is also possible that the upregulation of MT1-MMP by HIF-2 is specific to renal cell carcinoma, thereby providing an explanation as to why these VHL tumors are so invasive. Interestingly, VHL tumors differ in their expression of the HIF-α isoforms with some tumors types expressing exclusively one isoform and others both (Maxwell et al., 1999). Moreover, studies show that these two isoforms have limited overlap in their target gene profiles and exhibit cell-type specific expression with low redundancy (Giatromanolaki et al., 2001; Jain et al., 1998; Tian et al., 1997; Wiesener et al., 1998). Interestingly, inhibition of HIF-2α is both necessary and sufficient for tumor suppression by VHL in renal cell carcinoma in vivo (Kondo et al., 2003; Kondo et al., 2002a; Zimmer et al., 2004), whereas inhibition of HIF-1α is not (Maranchie et al., 2002). These findings suggest that HIF-2 behaves as a renal oncogene in the context of VHL disease. Normal epithelial cells do not express MT1-MMP (Seiki, 2003); therefore, given the role of MT1-MMP in cancer progression, we propose that HIF-2 induction of MT1-MMP expression may contribute to the oncogenic effects attributed to HIF-2 in RCC.
Our data show a cooperation of HIF-2 and Sp1 in the induction of MT1-MMP expression; however, other factors must contribute to HIF-2 activation of MT1-MMP gene expression given that mutations of both the functional HBS and Sp1 sites did not fully abrogate induced expression. One explanation for this result may be attributed to signaling through the chemokine receptor, CXCR4. CXCR4 is a hypoxia-inducible gene, and its upregulation due to the loss of VHL leads to increased invasiveness of RCC cells in response to its ligand, stromal-derived factor 1 (SDF-1) (Staller et al., 2003). Although the mechanism of how CXCR4 signaling enhances RCC tumor cell invasion has not been defined, a recent study in melanoma cells showed that the SDF-1/CXCR4 axis induces MT1-MMP expression, which was necessary for invasion (Bartolome et al., 2004). Whether VHL RCC tumor invasion is mediated by CXCR4 via the upregulation of MT1-MMP remains to be determined and is under current investigation.
Recently, Ueda et al. demonstrated that specific inhibition of MT1-MMP in tumor cells overexpressing MT1-MMP was sufficient to suppress cell migration and invasion (Ueda et al., 2003). Furthermore, Maquoi et al. showed in their recent studies that an orally active MMP inhibitor, selective for MT1-MMP, MMP-2, and MMP-9, prevented tumor cell invasion in vitro and had anti-tumor and anti-angiogenic activity in vivo (Maquoi et al., 2004). Our data define a direct link between the loss of the VHL tumor suppressor and the upregulation of MT1-MMP. Designing specific therapies, such as targeting MT1-MMP expression, may prove effective at preventing metastatic RCC disease and reduce the need for radical, invasive surgeries for VHL patients.
Materials and Methods
Cell lines and cell culture
All cell lines were maintained in Dulbecco’s Modified Eagle’s Medium (Life Technologies, Inc.) supplemented with 10% fetal bovine serum (Cambrex), penicillin 100U/mL, streptomycin 100ug/mL, and L-glutamine and cultured at 37°C, 5%CO2. The WT8 and pRc-9 cell lines (kindly provided by William Kaelin, Dana-Farber Cancer Institute, Boston, MA) were derived from the human 786-0 renal cell carcinoma cell line, which has a single VHL allele harboring a frameshift mutation at codon 104 resulting in a truncated, non-functional protein (Iliopoulos et al., 1995). 786-0 subclones stably transfected with pRc/CMV-HA-VHL (WT8) or pRc/CMV empty (pRc-9) were selected with the addition of 1mg/mL G418 sulfate. The WT8 cells were additionally stably transfected with pBabe-puro-HA-HIF2α, designated W+2PA (also provided by William Kaelin). pBabe-puro-HA-HIF2α contains two mutations (P405A, P531A), which abrogate prolyl-hydroxylation of HIF-2α, thereby inhibiting its interaction with VHL with no effect on the function of HIF-2α (Kondo et al., 2002a). The W+2PA cell line was selected with 1mg/mL G418 sulfate and 1.5μg/mL puromycin. HEK 293 cell line was purchased from American Type Culture Collection (ATCC). For endogenous gene expression experiments, cell lines were grown to confluence, washed 3 times in HBSS, and cultured in serum-free medium for 24 hours. For transfection studies, cell lines were transfected in serum-containing medium in the absence of antibiotics for 18 hours. MT1-MMP expression was not regulated by the presence or absence of serum (data not shown).
Reagents and plasmids
WP631 was obtained from Calbiochem. pGL3-Basic-0.4MT-luc, pGL3-Basic-0.4ΔSp1MT-luc, and pGL3-Basic-0.1MT-luc constructs have been previously described (Lohi et al., 2000). Briefly, these reporter constructs contain 0.4kb or 0.1kb of the 5’-flanking region of the MT1-MMP gene linked to the firefly luciferase gene. The 0.4MT-luc construct contains wild-type promoter sequence, whereas the 0.4ΔSp1MT-luc construct contains mutations in the functional Sp1 site at −92 which abrogate binding and transactivation by Sp1. The 0.1kb construct does not contain a functional Sp1 site.
MT1-MMP HBS mutant constructs were generated using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer’s protocol. The 0.4MT-luc promoter construct was used in the mutagenesis to create reporter plasmids which contained one or both of the putative HBSs mutated (Table 1). The oligos used in the mutagenesis were designed from the MT1-MMP sequence (GenBank accession #AF158733) using Stratagene’s web-based primer design software found at http://labtools.stratagene.com/QC . ΔHBS-1(−125) was created by mutating the sequence 5’-CACGTC-3’ to 5’-CTTTTC-3’. ΔHBS-2(+144) was created by mutating the sequence 5’-GGCGTG-3’ to 5’-GGAAAG-3’. Underlined nucleotides represent the mutated bases of the HBS consensus sequence. Mutations were confirmed by DNA sequence analysis. pTK-RE3-luc reporter plasmid contains 3 copies of a 50nt hypoxia-inducible enhancer from the erythropoietin gene and has been shown to be inducible by co-expression of HIF-2α (Tian et al., 1997). The pTK-RE3-luc, pCMV-HIF-2α, and pCMV-HIFβ expression constructs were generous gifts of Richard Bruick (University of Texas, Southwestern Medical Center, Dallas, TX). pRc/CMV-HA-VHL expression construct was a kind gift from William Kaelin. pRc/CMV empty vector was created from pRc/CMV-HA-VHL by HindIII/XbaI digest to remove the HA-VHL insert and was religated using HindIII-XmnI + XbaI-XmnI adaptors (NE Biolabs).
Northern blotting
Total cellular RNA was purified from using TRIzol Reagent (Invitrogen). Total RNA (10μg/lane) was separated by electrophoresis using 1% agarose formaldehyde gels and transferred to GeneScreen Plus membranes (NEN Lifesciences). Membranes were hybridized with a denatured MT1-MMP cDNA probe, labeled with α-32P dCTP by random oligo priming. Ethidium bromide stained 28S rRNA was used as a loading control.
Real-time RT-PCR
Total cellular RNA was purified from using either the TRIzol Reagent (Invitrogen) and subjected to DNase treatment (Ambion DNA-free kit) or the RNeasy kit (Qiagen) with on-column DNase treatment. Reverse transcription (RT) was performed using the Applied Biosystems Taqman Reverse Transcription Reagent Kit and following the manufacturer’s protocol. Briefly, 4μg of DNase treated RNA was used per 40μL reaction (5.5mM MgCl2, 500μM of each dNTP, 2.5μM oligod(T)16, 0.4U/μL RNase Inhibitor, 1.25U/μL Multiscribe enzyme). Alternatively, an MT1-MMP antisense primer, which is specific for exon 2, was used in the RT reactions in order to enrich for MT1-MMP RNAs. RT reactions were incubated at 25°C for 5 minutes, 48°C for 30 minutes, and 95°C for 5 minutes. Real-time PCR reactions were performing using the Applied Biosystems Sybr Green master mix. Five hundred ng of input cDNA was used in each real-time PCR reaction analyzing MT1-MMP expression, while 100 ng of input cDNA was used in each real-time PCR reaction analyzing GAPDH expression. Each cDNA was assayed in triplicate using a MJ Research DNA Engine Opticon thermal cycler using the following parameters: 95°C 10min, 40 cycles of 95°C 15s, 60°C 1min, and a plate read.
Standard curves were included in each assay. Standards for MT1-MMP mRNA and hnRNA were generated by serial log dilutions of the appropriate PCR product ranging from 10pg to 0.01pg, and 0.1pg to 0.0001pg respectively. Standards for GAPDH expression were generated using serial log dilutions ranging from 1ng to 0.001ng of the pCMV sport6 plasmid (ATCC). Primer sequences are as follows: MT1-MMP mRNA: 5’-CCCCGAAGCCTGGCTACA-3’ (sense) and 5’-GCATCAGCTTTGCCTGTTACT-3’ (antisense); MT1-MMP hnRNA: 5’-CTCTAAGCCATACCCCTTTCC-3’ (sense) and 5’-GCATCAGCTTTGCCTGTTACT-3’ (antisense); GAPDH mRNA: 5’-CGACAGTCAGCCGCATCTT-3’ (sense) and 5’-CCCCATGGTGTCTGAGCG-3’ (antisense). Data are presented as pg MT1-MMP per ng of GAPDH averaged from 3 or more individual experiments.
Immunoblotting
Whole cell lysates were harvested from confluent 6-well plates cultured in serum-free medium for 24 hours by washing the cells twice with cold 1x PBS, adding 150μL of lysis buffer (50mM Tris-HCl, pH 6.8, 1% SDS, 1mM EDTA, pH 8.0, protease inhibitor cocktail) or SDS reducing buffer (60mM Tris-HCl, pH 6.8, 2% SDS, 14.4mM β-mercaptoethanol, 25% glycerol, 0.1% bromphenol blue), and boiling for 5 minutes. Proteins were resolved by SDS-PAGE and electrotransferred to Immobilon-P PVDF membranes (Millipore). Membranes were blocked with 5% milk in Tris-buffered saline 0.1% Tween-20 at room temperature for 1–2 hours. Primary antibodies were diluted in blocking buffer and incubated with the membranes overnight at 4°C with rocking. Appropriate secondary antibodies were diluted in blocking buffer and incubated with the membrane at room temperature for 1 hour. Proteins were visualized by Western Lightning Chemiluminescence Reagent (Perkin Elmer). Antibodies and dilutions are as follows: VHL monoclonal antibody, 1:100 (Neomarkers, Clone Ig33); HA-HRP monoclonal antibody, 1:500 (Sigma); HIF-2α polyclonal antibody, 1:1000 (Novus Biologicals, NB100-122); actin monoclonal antibody, 1:5000 (Oncogene, Ab-1); anti-mouse HRP conjugated antibody, 1:5000 (Santa Cruz Biotechnology); and anti-rabbit HRP conjugated antibody, 1:2000 (Santa Cruz Biotechnology).
MT1-MMP ELISA
The Matrix Metalloproteinase-14 (MMP-14) Biotrak Activity Assay System (Amersham Biosciences), a quantitative measure of MT1-MMP activity, was used to analyze MT1-MMP protein expression. Cells were plated in serum-containing media in triplicate and harvested at confluence with the extraction buffer provided in the kit. The assay was conducted according to the manufacturer’s protocol. A standard curve ranging from 0.125ng/mL to 32ng/mL was used to quantitate protein expression. Because the assay is colorimetric and does not require quenching, the plate was read at 2.5hr, 6hr, and 9hr after incubation with the substrate. The data represent the 6hr timepoint since the values obtained at this time fit the standard curve most appropriately. Values are presented as average protein concentration normalized to average cell number at time of harvest (n=3).
Transient transfections
Cell lines were plated at a density of 2x105 cells/well in 6-well plates. When cells were 95% confluent, they were transiently transfected with 2–4μg of DNA using Lipofectamine 2000™ Transfection Reagent (Invitrogen) following manufacturer’s instructions. In co-transfection and expression construct dose response studies, empty vector controls were used to balance the concentration of transfected DNA. All transfections were performed in triplicate. Approximately 18hr later, cells were washed in cold 1x PBS and lysed using 25mM glycylglycine, 4mM EGTA, 15mM MgSO4, 1% Triton X-100 for luciferase assays or total RNA was harvested using the RNeasy kit as described under Real-time RT-PCR. Luciferase assays were run using a LMaxII384 Luminometer (Molecular Devices). Transfection efficiency was determined as a percentage of GFP-expressing cells counted from 3 fields of 3 separate transfections. Values are presented either as relative luciferase units (RLUs) of representative data or as mean control values from 3 or more experiments.
Statistical analysis
Statistical significance was calculated using the student’s t-test available at http://www.physics.csbsju.edu/stats/t-test.html and are represented as +/− standard deviation (S.D.) of the mean. Significance was assigned to p values <0.05.
Acknowledgments
This work was supported by National Institutes of Health grants AR-26599 (C.E.B) and CA-77267 (C.E.B.), by a grant from the Milheim Foundation for Cancer Research #2003–9 (B.L.P), and by the Academy of Finland and the Finnish Cancer Foundation (J.L). B.L.P. is supported by a pre-doctoral fellowship from NIH (T32-AI07363).
References
- Annabi B, Lee YT, Turcotte S, Naud E, Desrosiers RR, Champagne M, Eliopoulos N, Galipeau J, Beliveau R. Stem Cells. 2003;21:337–47. doi: 10.1634/stemcells.21-3-337. [DOI] [PubMed] [Google Scholar]
- Barry RE, Krek W. Trends Mol Med. 2004;10:466–72. doi: 10.1016/j.molmed.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Bartolome RA, Galvez BG, Longo N, Baleux F, Van Muijen GN, Sanchez-Mateos P, Arroyo AG, Teixido J. Cancer Res. 2004;64:2534–43. doi: 10.1158/0008-5472.can-03-3398. [DOI] [PubMed] [Google Scholar]
- Bogenrieder T, Herlyn M. Oncogene. 2003;22:6524–36. doi: 10.1038/sj.onc.1206757. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Werb Z. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- Gaidarova S, Jimenez SA. J Biol Chem. 2002;277:38737–45. doi: 10.1074/jbc.M201742200. [DOI] [PubMed] [Google Scholar]
- Giatromanolaki A, Koukourakis MI, Sivridis E, Turley H, Talks K, Pezzella F, Gatter KC, Harris AL. Br J Cancer. 2001;85:881–90. doi: 10.1054/bjoc.2001.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AL. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Hes FJ, van der Luijt RB, Lips CJ. Neth J Med. 2001;59:225–34. doi: 10.1016/s0300-2977(01)00165-6. [DOI] [PubMed] [Google Scholar]
- Hotary K, Allen E, Punturieri A, Yana I, Weiss SJ. J Cell Biol. 2000;149:1309–23. doi: 10.1083/jcb.149.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- Iida J, Wilhelmson KL, Price MA, Wilson CM, Pei D, Furcht LT, McCarthy JB. J Invest Dermatol. 2004;122:167–76. doi: 10.1046/j.0022-202X.2003.22114.x. [DOI] [PubMed] [Google Scholar]
- Iliopoulos O, Kibel A, Gray S, Kaelin WG., Jr Nat Med. 1995;1:822–6. doi: 10.1038/nm0895-822. [DOI] [PubMed] [Google Scholar]
- Jain S, Maltepe E, Lu MM, Simon C, Bradfield CA. Mech Dev. 1998;73:117–23. doi: 10.1016/s0925-4773(98)00038-0. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. J Biol Chem. 1996;271:17771–8. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr Nat Rev Cancer. 2002;2:673–82. doi: 10.1038/nrc885. [DOI] [PubMed] [Google Scholar]
- Kitagawa Y, Kunimi K, Uchibayashi T, Sato H, Namiki M. J Urol. 1999;162:905–9. doi: 10.1097/00005392-199909010-00088. [DOI] [PubMed] [Google Scholar]
- Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr Cancer Cell. 2002a;1:237–46. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Kondo S, Kubota S, Shimo T, Nishida T, Yosimichi G, Eguchi T, Sugahara T, Takigawa M. Carcinogenesis. 2002b;23:769–76. doi: 10.1093/carcin/23.5.769. [DOI] [PubMed] [Google Scholar]
- Koochekpour S, Jeffers M, Wang PH, Gong C, Taylor GA, Roessler LM, Stearman R, Vasselli JR, Stetler-Stevenson WG, Kaelin WG, Jr, Linehan WM, Klausner RD, Gnarra JR, Vande Woude GF. Mol Cell Biol. 1999;19:5902–12. doi: 10.1128/mcb.19.9.5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehti K, Lohi J, Valtanen H, Keski-Oja J. Biochem J. 1998;334(Pt 2):345–53. doi: 10.1042/bj3340345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan WM, Walther MM, Zbar B. J Urol. 2003;170:2163–72. doi: 10.1097/01.ju.0000096060.92397.ed. [DOI] [PubMed] [Google Scholar]
- Lohi J, Lehti K, Valtanen H, Parks WC, Keski-Oja J. Gene. 2000;242:75–86. doi: 10.1016/s0378-1119(99)00549-1. [DOI] [PubMed] [Google Scholar]
- Maher ER, Kaelin WG., Jr Medicine (Baltimore) 1997;76:381–91. doi: 10.1097/00005792-199711000-00001. [DOI] [PubMed] [Google Scholar]
- Maquoi E, Sounni NE, Devy L, Olivier F, Frankenne F, Krell HW, Grams F, Foidart JM, Noel A. Clin Cancer Res. 2004;10:4038–47. doi: 10.1158/1078-0432.CCR-04-0125. [DOI] [PubMed] [Google Scholar]
- Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. Cancer Cell. 2002;1:247–55. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- Martin B, Vaquero A, Priebe W, Portugal J. Nucleic Acids Res. 1999;27:3402–9. doi: 10.1093/nar/27.17.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- McCawley LJ, Matrisian LM. Curr Opin Cell Biol. 2001;13:534–40. doi: 10.1016/s0955-0674(00)00248-9. [DOI] [PubMed] [Google Scholar]
- Miki N, Ikuta M, Matsui T. J Biol Chem. 2004;279:15025–31. doi: 10.1074/jbc.M313186200. [DOI] [PubMed] [Google Scholar]
- Mori H, Tomari T, Koshikawa N, Kajita M, Itoh Y, Sato H, Tojo H, Yana I, Seiki M. Embo J. 2002;21:3949–59. doi: 10.1093/emboj/cdf411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottino P, Finley J, Rojo E, Ottlecz A, Lambrou GN, Bazan HE, Bazan NG. Mol Vis. 2004;10:341–50. [PubMed] [Google Scholar]
- Richards FM, Webster AR, McMahon R, Woodward ER, Rose S, Maher ER. J Intern Med. 1998;243:527–33. doi: 10.1046/j.1365-2796.1998.00334.x. [DOI] [PubMed] [Google Scholar]
- Sanchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C. J Biol Chem. 2002;277:43799–808. doi: 10.1074/jbc.M207160200. [DOI] [PubMed] [Google Scholar]
- Sanchez-Elsner T, Ramirez JR, Sanz-Rodriguez F, Varela E, Bernabeu C, Botella LM. J Mol Biol. 2004;336:9–24. doi: 10.1016/j.jmb.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, Seiki M. Nature. 1994;370:61–5. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- Seiki M. Cancer Lett. 2003;194:1–11. doi: 10.1016/s0304-3835(02)00699-7. [DOI] [PubMed] [Google Scholar]
- Singh AD, Shields CL, Shields JA. Surv Ophthalmol. 2001;46:117–42. doi: 10.1016/s0039-6257(01)00245-4. [DOI] [PubMed] [Google Scholar]
- Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Nature. 2003;425:307–11. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI. J Biol Chem. 1995;270:5331–8. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- Tian H, McKnight SL, Russell DW. Genes Dev. 1997;11:72–82. doi: 10.1101/gad.11.1.72. [DOI] [PubMed] [Google Scholar]
- Ueda J, Kajita M, Suenaga N, Fujii K, Seiki M. Oncogene. 2003;22:8716–22. doi: 10.1038/sj.onc.1206962. [DOI] [PubMed] [Google Scholar]
- Visse R, Nagase H. Circ Res. 2003;92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, Ratcliffe PJ, Maxwell PH. Blood. 1998;92:2260–8. [PubMed] [Google Scholar]
- Zimmer M, Doucette D, Siddiqui N, Iliopoulos O. Mol Cancer Res. 2004;2:89–95. [PubMed] [Google Scholar]