

Abstract
A significant amount of attention has recently been focused on modeling of gene regulatory networks. Two frequently used large-scale modeling frameworks are Bayesian networks (BNs) and Boolean networks, the latter one being a special case of its recent stochastic extension, probabilistic Boolean networks (PBNs). PBN is a promising model class that generalizes the standard rule-based interactions of Boolean networks into the stochastic setting. Dynamic Bayesian networks (DBNs) is a general and versatile model class that is able to represent complex temporal stochastic processes and has also been proposed as a model for gene regulatory systems. In this paper, we concentrate on these two model classes and demonstrate that PBNs and a certain subclass of DBNs can represent the same joint probability distribution over their common variables. The major benefit of introducing the relationships between the models is that it opens up the possibility of applying the standard tools of DBNs to PBNs and vice versa. Hence, the standard learning tools of DBNs can be applied in the context of PBNs, and the inference methods give a natural way of handling the missing values in PBNs which are often present in gene expression measurements. Conversely, the tools for controlling the stationary behavior of the networks, tools for projecting networks onto sub-networks, and efficient learning schemes can be used for DBNs. In other words, the introduced relationships between the models extend the collection of analysis tools for both model classes.
Keywords: Gene regulatory networks, Probabilistic Boolean networks, Dynamic Bayesian networks
1. Introduction
During recent years, it has become evident that cellular processes are executed in a highly parallel and integrated fashion and that computational modeling approaches can provide powerful methodologies for gaining deeper insight into the operation of living cells. The modeling problem that has received a considerable amount of attention is the discovery of transcriptional level interactions. With the help of recent development in high-throughput genomic technologies, computational methods have enormous potential in the context of model inference from real measurement data and in practical use, such as drug discovery.
A number of different frameworks for gene regulatory network modeling have been proposed, ranging from differential equations too qualitative models (for an overview, see e.g. [1]). There is a clear conceptual difference between differential equation and coarse-scale models. The former can be used for a detailed representation of biochemical reactions, whereas the latter emphasize fundamental, generic principles between interacting components. In this context, models classes that are both discrete-time and discrete-state are called coarse-scale models.
Fine-scale modeling of biological interactions at the molecular level may require some type of differential equations. Although differential equations have successfully been used to simulate small (known) biochemical pathways (see e.g. [2,3]), their use in large-scale (genome-wide) modeling has considerable limitations. First of all, those models are computationally very demanding. Therefore, when modeling regulatory networks with differential equations, the model selection problem is usually ignored and the underlying biological system is assumed to be known. Because the model selection is the most important computational tool for discovering new, unknown regulatory relationships from the measured data, researchers have considered alternative modeling approaches. Also, the available analysis tools for differential equations are much more restricted than the ones for the alternative model classes (see below).
So-called graphical models can overcome the above-mentioned modeling problems, and advanced analysis tools have been developed for them. The use of holistic, coarse-scale models is also supported by the fact that the currently available data is limited both in quality and the number of samples. That is, there is no advantage using models that are much more accurate than the available data. Another constraint to be kept in mind is that the modeling framework should also be selected on the basis of the preferred goals, i.e., to what kinds of questions are we seeking answers. The two most often used large-scale modeling frameworks are Boolean and Bayesian networks (BNs). Since the Boolean network is a special case of another commonly used model class, probabilistic Boolean networks (PBNs), we will consider PBNs instead of Boolean networks.
PBNs is a model class that has been recently introduced in the context of genetic network modeling [4]. PBN is a stochastic extension of the standard Boolean network that incorporates rule-based dependencies between variables but is also stochastic in nature. The PBN model has a strong biological motivation through the standard, often used Boolean network model, originally proposed by Kauffman [5,6]. The theory of PBNs as models of genetic regulatory networks has been developed further in several papers. In particular, there has been interest in the control of stationary behavior of the network by means of gene interventions/perturbations [7], modifications of the network structure [8], and external control [9]. Another recent paper [10] introduces mappings between PBNs, including projections, node adjunctions and resolution reductions, which at the same time preserve consistency with the original probabilistic structure. Further, learning methods for PBNs have been introduced in [11,12]. More efficient learning schemes, in terms of computational complexity, but with cost of decreased accuracy, have been studied in [13]. General learning concepts have also been introduced in [14], although not in the context of PBNs, but a related setting. Kim and co-authors also show that the Markovian gene regulatory network model1 is biologically plausible [15].
Dynamic Bayesian networks (DBNs), also called dynamic probabilistic networks, are a general model class that is capable of representing complex temporal stochastic processes [16–18]. DBNs are also known to be able to capture several other often used modeling frameworks, such as hidden Markov models (and its variants) and Kalman filter models, as its special cases (see e.g. [18]). DBNs and their non-temporal versions, BNs, have successfully been used in different engineering problems, such as in speech recognition [19] and target tracking and identification [20]. Recently, BNs have also been used in modeling genetic regulation [17,21–30].
In this study we concentrate on PBNs and DBNs, and introduce certain equivalences between them. The first part of this paper is devoted to showing that PBNs and a certain subclass of DBNs can represent the same joint probability distribution over their common variables. For that purpose, we introduce a way of conceptually expressing a PBN as a DBN and vice versa. We would like to note that because there are many PBNs that can represent the same conditional probabilities, the one-to-one connection between the two models is true only in terms of probabilistic behavior. The main motivation for introducing the relationships between the models is that it opens up the possibility of applying the advanced tools of these network models to both of them. In other words, the introduced relationships between the models extend the collection of analysis tools for both model classes. The most important consequences of the results are briefly summarized below.
From the DBN point of view, the tools for controlling the stationary behavior of PBNs, by means of interventions, structural modifications of the network, and optimal external control, become available for DBNs. To our knowledge, no such methods have been introduced in the context of DBNs thus far. The same applies to efficient learning schemes, as well as mappings between different networks, in particular, projections onto subnetworks, which at the same time preserve consistency with the original probabilistic structure.
From the PBN point of view, one can use the standard learning tools of DBNs. This is particularly useful because the learning of gene regulatory networks has turned out to be a difficult problem and, therefore, efficient and flexible tools, with a possibility to be able to combine different data sources, are needed. Furthermore, both exact and approximate inference tools give a natural way of handling the missing values in PBNs which are often present in gene expression measurements. Further discussion on the impacts of the relationships can be found in Section 6. Note that the presented results are applicable to all similar modeling approaches, not only for gene regulatory network modeling. Also note that although the relationships are presented in the binary setting, they can be generalized to finer models (more discretization levels) as well.
The paper is organized as follows. Section 2 covers the basics of both model classes and develops PBNs to the extent necessary for this paper. Sections 3 and 4 introduce the relationships between the two models while the benefits of the relationships are explained in Section 6. Section 7 is devoted to general discussion and further topics in learning gene regulatory networks.
2. Network models
In the following, we focus on distributions over a set of discrete-valued random variables. To make a distinction between random variables and their particular values we use the following notation. Upper-case letters, such as X, X1, Y, are used to denote random variables (and corresponding nodes in the graphs). Lower-case letters, such as x, x1, y, are used to denote the values of the corresponding random variables. Vector-valued quantities are in boldface.
2.1. Probabilistic Boolean networks
For consistency of notation, we will be using the same notation as in [4]. A PBN G(V, F) is defined by a set of binary-valued nodes (genes) V = {X1, …, Xn} and a list of function sets F = (F1, …, Fn), where each function set Fi consists of l(i) Boolean functions, i.e., . The value of each node Xi is updated by a Boolean function taken from the corresponding set Fi. A realization of the PBN at a given time instant is defined by a vector of Boolean functions. Assuming that there are N possible realizations for the PBN, then there are N vector functions f1, …, fN where each , 1≤j≤N, 1 ≤ ji ≤l(i), and each . Each realization of the PBN maps (updates) the values of the nodes into their new values, i.e., fj : 𝔹n → 𝔹n, where 𝔹 = {0, 1}. Network realizations f1, …, fN constitute the possible values of a random variable whose outcome is selected independently for each updating step.
In order to make the discussion more explicit, we use the notion of time with the updating step of the network. That is, X i(t) (1≤i≤n) is the discrete-time random variable (stochastic process) that denotes the attribute X i at time t, and X(t) = (X1(t), …, X n(t)) is a vector of all random variables X i, 1≤i≤n, at time t. So, the updating step from time t − 1 to t, given the current state of the nodes x(t −1) and a realization fj, is expressed as (x1(t), …, xn(t)) = fj(x1(t −1), …, xn(t −1)).
Let F(i) and F = (F(1), …, F(n)) denote random variables taking values in and F1 × … × F n, respectively. The probability that a certain predictor function is used to update the value of the node X i is equal to
| (1) |
A PBN is said to be independent if the elements F(1), …, F(n) of the random variable F are independent, i.e., . If the PBN is independent, its predictor functions , 1≤j≤l(i), for all the nodes X i (1≤i≤n), are associated with an independent selection probability . A PBN is said to be dependent if the joint probability distribution of random variables F(1), …, F(n) cannot be factored as for the independent PBN. Correspondingly, the node X i in the PBN is said to be independent (resp. dependent) if its predictor function is (resp. is not) independent of the other predictor functions. It is also worth noting that the model G(V, F) is not changing with time, i.e., it defines a homogeneous process. A basic building block of a PBN, which describes the updating mechanisms for a single node Xi, is shown in Fig. 1.
Fig. 1.
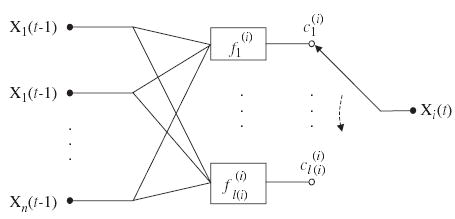
A basic building block of probabilistic Boolean networks (PBN). The graph describes the updating mechanisms for a single node Xi in the network.
2.2. Time-series in PBNs
In the following, we are interested in the joint probability distribution of the variables expanded over a finite number of updating steps, say T steps, i.e., {X(i) : 0≤i≤T}. Given the initial state x(t − 1), the probability of moving to some state x(t) after one step of the network is (see e.g. [4])
| (2) |
Because the network realizations are selected independently for each time instant, the joint probability distribution over all possible time-series of length T + 1 can be expressed as
| (3) |
where Pr{X(0) = x(0)} denotes the distribution of the first state. Eq. (3) shows that dynamics of PBNs can also be modeled by Markov chains [4].
Let us concentrate on independent PBNs for now and rewrite the state transition probabilities A(x(t − 1), x(t)). Let (x(t))i, 1≤i≤n, denote the ith element of x(t), and A(x(t −1), (x(t))i) denote the probability that the ith element of x(t) will be (x(t))i after one step of the network, given that the current state is x(t −1). (A more detailed computation of probability A(x(t −1), (x(t))i) is shown in Eq. (12), which will be discussed later on.) Since all nodes are assumed to be independent, each node is updated independently, and Eq. (2) can be written as [15]
| (4) |
So, given Eqs. (3) and (4), the joint probability distribution over all possible time-series of length T + 1 can be expressed as
| (5) |
Let us now concentrate on a dependent PBN. Let a dependent PBN have I independent nodes, X i, i = 1, …, I, and D sets of dependent but mutually exclusive nodes, Xj = (Xj1,…, Xjd(j)), j = 1, …, D. That is, {X i} ∩ Xj = ∅;, for all i = 1, …, I, j = 1, …, D, and Xj ∩ Xk = ∅, for all 1≤j≠k≤D.2 Further, . Without loss of generality, we assume that the genes are sorted and re-labeled such that the first I nodes are the independent ones and the rest are dependent. The probability distribution of the network realization can be written as
| (6) |
Define Dj = {j1, …, jd(j)}, and let (x(t))Dj denote the elements j1, …, jd(j) of the vector x(t). Let A(x(t − 1), (x(t))Dj) denote the probability that the elements j1, …, jd(j) of x(t) are (x(t))Dj after one step of the network, given that the current state is x(t − 1). Without going into details, it is quite straightforward to see that the joint probability of a dependent PBN over all T +1 length time-series can be decomposed the same way as in Eq. (5) except that terms A(x(t −1), (x(t))i), which correspond to the independent nodes, must be replaced by terms A(x(t −1), (x(t))Dj). That is,
| (7) |
2.3. Dynamic Bayesian networks
Definitions in this section follow mainly the notation used in [17]. Let X = {X1, …, Xn} denote the (binary-valued) random variables in the network and Pr{·} denote the joint probability distribution of X.3 A BN, also called a probabilistic network, for X is a pair B = (G, Θ) that encodes the joint probability distribution over X. The first component, G, is a directed acyclic graph whose vertices correspond to the variables in X. The network structure, especially the lack of possible arcs in G, encodes a set of conditional independence properties about the variables in X. The second component, Θ, defines a set of local conditional probability distributions (or conditional probability tables (CPT)), induced by the graph structure G, associated with each variable. Let Pa(Xi) denote the parents of the variable Xi in the graph G and pa(Xi) denote the value of the corresponding variables. Then, a BN B defines a unique joint probability distribution over X given by
| (8) |
For a detailed introduction to BNs, see, e.g., [31,32].
Temporal extension of BNs, DBNs, extend these concepts to stochastic processes. In this paper, we restrict our attention to first-order Markov processes in X, i.e., to processes whose transition probability obeys Pr{X(t)|X(0), X(1), …, X(t − 1)} = Pr{X(t)|X(t − 1)}. The transition probabilities are also assumed to be independent of t, meaning that the process is homogeneous, as is the case for PBNs.
A DBN that represents the joint probability distribution over all possible time-series of variables in X consists of two parts: (i) an initial BN B0 = (G0, Θ0) that defines the joint distribution of the variables in X(0), and (ii) a transition BN B1 = (G1, Θ1) that specifies the transition probabilities Pr{X(t)|X(t − 1)} for all t. So, a DBN is defined by a pair (B0, B1). In this paper we restrict the structure of DBNs in two ways. First, the directed acyclic graph G0 in the initial BN B0 is assumed to have only within-slice connections, i.e., Pa(Xi(0)) ⊆ X(0) for all 1≤i≤n. We also constrain the variables in time slice X(t), t>0, to have all their parents in slice t − 1, i.e., Pa(Xi(t)) ⊆ X(t − 1) for all 1≤i≤n and t>0. The fact that connections exist only between consecutive slices is related to our first-order Markovian assumption stated earlier. An example of the basic building blocks of DBNs, B0 and B1, is shown in Fig. 2.
Fig. 2.
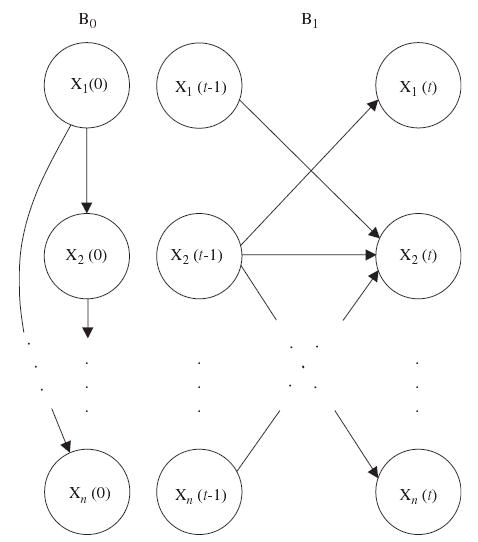
An example of the basic building blocks of dynamic Bayesian networks (DBNs) (B0, B1). B0 and B1 are the initial and transition Bayesian networks (BNs), respectively.
Using Eq. (8) and the assumptions on the initial and transition BNs discussed above, the joint distribution over a finite set of random variables X(0) ∪ X(1) ∪ … ∪ X(T) can be expressed as [17]
| (9) |
It may also be worth mentioning that we assume fully observable DBNs. That is, when used in a real application, the values of all nodes are observed. Hidden nodes are considered when extensions to PBNs are discussed in Section 5.
3. Relationships between independent PBNs and DBNs
In order to be able to establish the relationship between PBNs and DBNs, we will add an extra feature to the definition of a PBN. We will assume that the initial state X(0) of a PBN can have any joint probability distribution. Indeed, that definition was already used in Eqs. (3), (5) and (7). In particular, we assume X(0) to have the same distribution as defined by B0 for the first slice of a DBN. For instance, in the context of genetic regulatory network modeling this initial distribution can be set equal to the stationary distribution of the corresponding Markov chain. Also note that we are not discussing the general class of DBNs in the following but only the subclass of binary-valued DBNs, as discussed in the previous section. We relax the requirement of binary-valued nodes in Sections 4.1 and 4.2.
3.1. An independent PBN as a binary-valued DBN
We first illustrate a way of expressing an independent PBN G(V,F) as a DBN (B0,B1). Let an independent PBN G(V,F) be fixed. The nodes in the graphs G0 and G1 must clearly correspond to the nodes in the PBN. In order to distinguish between the nodes from different models, nodes in the PBN are denoted by X i, 1≤i≤n, as above, and the corresponding nodes in the DBN are denoted by (similarly for the vector-valued random variables X and ). We refer to nodes X i and X as the PBN counterparts of and , respectively, and the other way around.
Because an initial BN B0 is capable of representing any joint probability distribution over its nodes [31,32], the distribution of the initial state of the PBN, Pr{X(0)}, can be represented by B0. We omit the technical details since they are quite straightforward.
From Eqs. (3) and (9) it is easy to see that both PBNs and DBNs obey the first-order Markovian property. Thus, we only need to consider one-step transition probabilities, say between time instants t − 1 and t, Pr{X(t)|X(t − 1)}, when expressing the PBN as a DBN. Further, from Eqs. (5) and (9) one can see that, for both models, the joint probability can also be decomposed over their nodes in the same way. Thus, when constructing a transition BN B1, we can further concentrate only on a single node, say X i, with other nodes being handled similarly. Let denote the set of essential variables (nodes) used by predictor function for gene X i at time t. The set
| (10) |
denotes the set of all variables used to predict the value of the gene Xi. Let us expand the domain of all the predictor functions in Fi by adding fictitious variables so that they are functions of variables in Pa(Xi(t)). Thus, we can define B1 = (G1,Θ1) as follows. The graph G1 has directed edges from to such that the parents of are equal to the DBN counterparts of the nodes shown in Eq. (10). As was already shown in [4], given the distribution 𝒟i over the predictor variables of node X i (denoted as Pa(X i)~ 𝒟i), the probability of X i being one is
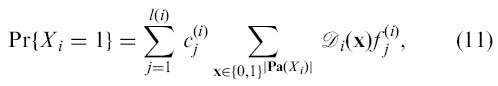
where the domain of predictor functions is assumed to be expanded and is treated as a real-valued function. We can interpret Eq. (11) as Pr{X i(t) = 1|Pa(Xi(t))~ 𝒟i}. Thus, by specifying 𝒟i to be “deterministic” such that it corresponds to a particular predictor node configuration Pa(X i(t)) = z, i.e., 𝒟i(x) = 1 if x = z and 𝒟i(x) = 0 otherwise, we have that
| (12) |
Then, the set of local conditional probability distributions, or CPTs, Θ1, induced by the graph structure G1, has exactly the same entries as shown in Eq. (12) for each node. So, all terms in Eq. (5) can be represented by corresponding terms in Eq. (9). Thus, any PBN can be expressed as a binary DBN.
3.2. A binary-valued DBN as an independent PBN
To establish the converse of the above conclusion, let us see how a binary-valued DBN (B0,B1) can be represented as an independent PBN G(V,F). Let a DBN (B0,B1) be given. The set of nodes V must clearly correspond to the nodes in the graphs G0 and G1, and the distribution of the first state X(0) in the PBN must be the same as for the DBN.
Following the same reasoning as in Section 3.1, we can again conclude that when constructing a PBN, one only needs to consider the predictor functions for a single node X i between consecutive time instants t − 1 and t. Let us assume that the local conditional probability distributions in Θ1 are given in the form of CPTs, and the number of parents of the ith node is denoted as Let us enumerate the entries in the CPTs that are assigned to the ith node as triplets (zl,yl,pl), where zl ∈ 𝔹, yl ∈ 𝔹q, and 1 ≤l≤2q+1. Let us also suppose that the triplets are enumerated such that the first r = 2q triplets have zl = 1 and they are sorted in increasing order, i.e., 1≤k<l≤r ⇒ pk≤pl. Let yl = yl1yl2 … ylq and interpret a sequence of symbols as a conjunction, where if yli = 1 and if yli = 0. The variables in the conjunction correspond to a set of specific variables, {Xl1,…, Xlq}, in the PBN, i.e., they are PBN counterparts of
Then, for the ith node in the PBN, the set of functions F i can be generated as follows: where
| (13) |
is a disjunction of conjunctions, is a zero function, i.e., for all x ∈ 𝔹q, and r = 2q.4 The corresponding selection probabilities are set to for 2≤l≤r, and . If some of the happen to be zero, the corresponding functions can be removed from F i. By applying Eq. (12), we can verify that the above construction really gives an equivalent PBN to the given DBN. Let us first compute the one step prediction probabilities for the cases where X i(t) = 1 and the parent nodes have value yl. One gets
| (14) |
where the second equality follows from the fact that only the first l functions contain the conjunction xl (see Eq. (13)), i.e., only for 1 ≤j≤l. The probability of the corresponding complement event Pr{X i(t) = 0|Pa(X i(t)) = yl} is clearly 1 − pl. So, a binary-valued DBN can be represented as an independent PBN. Thus, we can state the following theorem.
Theorem 1
Independent PBNs G(V,F) and binary-valued DBNs (B0,B1) whose initial and transition BNs B0 and B1 are assumed to have only within and between consecutive slice connections, respectively, can represent the same joint distribution over their common variables.
The methods introduced in Sections 3.1 and 3.2 provide the actual conversions between the two modeling frameworks.
It is important to note that the mapping from a binary DBN to an independent PBN is not unique. Instead, there are many PBNs that have the same probabilistic structure. This is best illustrated by a toy example (see also the example below). Assume that node X1 is regulated by node X2 and all the values in the CPT of the node X1 are equal to 0.5. Then the following two function sets have the same conditional probabilities: , with and where the functions and are constant zero and unity functions, respectively, or , with and where the functions and are the identity and the negation functions, respectively. In other words, two fundamentally different function sets can have the same probabilistic structure. In the case of many parent variables, this issue becomes more complicated. In practice, one may want to construct the predictor functions for each node in the PBN such that the predictor functions have as few variables as possible, or such that the number of predictor functions is minimized.
Let us first consider minimizing the number of variables in the predictor functions. The above construction method produces predictor functions with the maximal number of variables. In some cases, when the CPTs are “separable,” one can construct predictor functions having less variables, but at the same time being consistent with the original conditional probabilities. For example, consider a case where node X1 is regulated by a set of nodes X2,…,Xn. Assume that the first parent node X2 has a forcing (canalizing) effect on the target node X1 such that all the values in the CPT for which X1 = 1 and X2 = 1 are equal to 0.9. Assume further (for simplicity) that all other values in the CPT for which X1 = 1 (i.e., X1 = 1 and X2 = 0) are smaller than 0.9, the largest of those being, say, 0.8. Instead of blindly using the aforementioned algorithm for generating the predictor functions for X 1, it can be useful to take the special form of the CPT into account. Following the above algorithm the first functions can be constructed as explained. However, the effect of the forcing variable X 2 can be accounted for using only a single one-variable predictor function f (1) = x2 with c(1) = 0.9 − 0.8 = 0.1. Alternatively, if no single-variable predictor functions can be constructed as explained above, then combinations of variables, starting with two variables, three variables, etc., can be considered. For example, if all the values in the CPT for which X 1 = 1, X 2 = 1 and X 3 = 1 are equal, then a single two-variable function can be defined as f (1) = x2x3.
The above search for predictor variables was explained in terms of the original conditional probabilities (i.e., CPTs). Alternatively, one can try to optimize the obtained predictor functions somehow. That is, each function in F i should be expressed in some optimal form. The first thing to be considered is the removal of fictitious variables from the functions. That can possibly result in functions which have far fewer input variables. Yet another issue is to further optimize the actual expressions of the Boolean functions using methods such as Quine–McCluskey algorithm (see e.g. [33]), which minimizes the number of terms in the disjunctive normal form. The above discussion, however, does not provide any optimal method for the predictor function construction (apart from the optimal representation of the functions). An optimal method can be described in terms of the number of predictor functions which is described next.
In the worst case, the minimum number of predictor functions is determined by the number of different values in the CPT. The above construction method automatically selects that number of predictors (plus a possible constant zero function). In some special cases, the number of predictor functions can be reduced. Consider again the same triplets as above (zl,yl,pl) for a single node X i and again focus on only those triplets for which zl = 1. This leaves us with a set of probability values p1,…,pr, r = 2q. The general optimality criterion can be stated as follows. Find the smallest set of selection probabilities , where each and , such that each pl, 1 ≤l≤r, can be written as
| (15) |
where Il ⊆ {1, …,m}. Let Jj = {l|j ∈ Il} ⊆ {1, …,2q} be the set of indices of those Pl for which is used in the sum in Eq. (15). Then the function set is defined as
where xl is as above. In the same manner as in Eq. (14), we can verify the correctness of the predictor functions and the corresponding selection probabilities
| (16) |
because (the third equality) only if the function contains the conjunction xl, i.e.,l ∈ Jj. The second to last equality follows from the fact that l ∈ Jj ⇔ j ∈ Il, and the last equality from Eq. (15). Note that the optimal function set can still be non-unique.
The obtained functions in Fi can be modified further as explained above e.g. by removing the possible fictitious variables and by applying the Quine–McCluskey optimization algorithm. A computationally efficient algorithm for the search of the optimal function sets remains to be developed. Fortunately, the search problem is usually limited in the sense that each gene contains only few parent variables.
Theorem 1 says that the two model classes can represent the same probabilistic behavior. However, there are many statistically equivalent PBNs that correspond to a DBN. This means the PBN formalism is redundant from the probabilistic point of view. On the other hand, PBN formalism is richer from the functional point of view because it can explain the regulatory roles of different gene sets in more detail than the conditional probabilities in DBNs can do.
Example 2
This example illustrates the above conversions between PBNs and DBNs. Let us assume that we have the following independent PBN: G = (V,F), V = {X1,X2}, F = (F1,F2), which we would like express as a DBN. (We omit the definition of the joint probability distribution of the first slice X(0).) The sets of essential variables used by predictor functions and are and , respectively. Then, following Eq. (10), the parents of the node are the DBN counterparts of . In a similar manner, we obtain that . For purposes of illustration, the truth tables of the functions and , as well as and , are shown in Table 1. Note that the domains of the functions and are expanded.
Table 1.
The truth tables of the functions and
| (x1x2) | ||||
|---|---|---|---|---|
| 00 | 1 | 0 | 0 | 1 |
| 01 | 1 | 1 | 0 | 1 |
| 10 | 0 | 0 | 0 | 0 |
| 11 | 1 | 1 | 1 | 0 |
Note that the domains of the functions and are expanded.
The local conditional probability distributions in the DBN can be obtained by using Eq. (12). For instance, the probability of being one, given that is . The probability of the corresponding complement event is Similar calculations apply to other cases. The local conditional probability distributions are tabulated in Table 2 which concludes the conversion of the given PBN into a DBN.
Table 2.
The local conditional probability distributions for the node and
| 1 | 00 | ||
| 1 | 01 | ||
| 1 | 10 | 0 | 0 |
| 1 | 11 | ||
| 0 | 00 | 1 − 0.2 = 0.8 | 1 − 0.4 = 0.6 |
| 0 | 01 | 1 − 1 = 0 | 1 − 0.4 = 0.6 |
| 0 | 10 | 1 − 0 = 1 | 1 − 0 = 1 |
| 0 | 11 | 1 − 1 = 0 | 1 − 0.6 = 0.4 |
Let us now assume that we are given a DBN as follows. (We again omit the definition of the initial BN B0.) The transition BN B1 = {G1, Θ1} is: G1 = {V1, E1}, with and The CPTs for both nodes (Θ1) are given in Table 2. Let us concentrate on the node and enumerate the triplets 1 (yl,zl,pl),1 ≤l≤ 8, in increasing order as introduced above (see Table 3).
Table 3.
The enumeration of the triplets (yl, zl, pl) for both nodes and
| (1, 10, 0) | (1, 10, 0) |
| (1, 00, 0.2) | (1, 00, 0.4) |
| (1, 01, 1) | (1, 01, 0.4) |
| (1, 11, 1) | (1, 11, 0.6) |
The enumeration is shown only for those triples for which yl = 1.
The function set F1 contains functions and , where the last one denotes the zero function. The corresponding selection probabilities are and . Because , the corresponding functions can be removed from F1. Further, the remaining functions can be manipulated as and which directly correspond to the predictor functions shown in the beginning of this Example. Note that the selection probabilities are also the same. After similar operations for the second node one gets the following list of functions: and , with the corresponding selection probabilities being and . These functions do not directly correspond to the ones shown in the beginning of this Example. However, in light of Eq. (12), they are effectively the same because they define the same one-step element-wise prediction probabilities (see also the last column of Table 2).
Note that the number of predictor functions can be reduced for the second node. Using the optimal procedure explained above it is easy to see that, e.g., the following two functions suffice , with and , where and .
4. Relationships between dependent PBNs and DBNs
This section shows the relationships between dependent PBNs and DBNs. The methods of expressing a dependent PBN as a DBN, and vice versa, are conceptually similar to the ones for independent PBNs.
4.1. A dependent PBN as a discrete-valued DBN
Let us concentrate on a dependent PBN and illustrate a way of expressing it as a DBN. Using the same notation as in Section 2.2, let the given dependent PBN have I independent nodes, Xi, i = 1, …, I, and D sets of dependent and mutually exclusive nodes, Xj = {Xj1, …, Xjd(j)}, j = 1, …, D. Assuming that the genes are sorted and re-labeled as in Section 2.2, the probability distribution of the network realization can be written as in Eq. (6).
From now on, we relax the requirement of pure binary-valued nodes by allowing discrete-valued nodes in DBNs. In the following the discrete-valued nodes are also thought of as binary vector-valued nodes (i.e., binary vector presentation of a discrete variable). In order to define an equivalent DBN to the given PBN, we start by defining its nodes. First, a DBN is assumed to have only I + D nodes. Let the first I nodes be binary-valued nodes as above, 1 ≤i≤ I, and the remaining D nodes be binary vector-valued, denoted as 1 ≤ j ≤ D, and X̂j ∈ 𝔹jd(j). The scalar- and vector-valued nodes, and correspond to the nodes Xi and Xj1, …, Xjd(j) in the PBN, respectively (see Fig. 3). As before, nodes Xi and Xj1, …, Xjkare called the PBN counterparts of and respectively. (Similarly, and are the DBN counterparts of Xi and Xj.) In vector form, the nodes in a DBN, when incorporating the notion of time, are
Fig. 3.
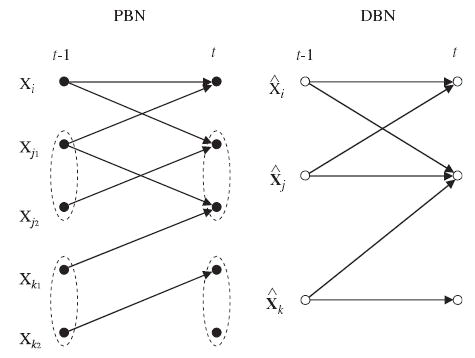
Correspondence between the nodes in a dependent PBN and a DBN (see the text for details).
An initial BN B0 can be defined as in Section 3.1, such that the nodes (1 ≤i≤I) and the elements of the nodes (1 ≤j≤D) in the DBN have the same initial joint distribution as the variable X(0) in the given PBN. Technical details are again omitted.
Let us then construct the transition BN B1 = (G1, Θ1). Following the same reasoning as in Section 3.1, the dependent PBN defines a first-order Markovian process in X(t) (see also Eq. (7)). Even though the given PBN is dependent, nodes of a DBN are constructed in such a way that their PBN counterparts are mutually independent (see Eq. (6)). Based on Eq. (7) and the above node construction, we can again concentrate on a single node in the DBN, between consecutive time steps t − 1 and t, when constructing the transition BN B1.
The set Pa(Xi(t)) denotes the set of all variables used to predict the value of the gene Xi at time t (see Eq. (10)). Similarly, let contain the DBN counterparts of the nodes in Pa(Xi(t)) except that variables belonging into sets of dependent nodes Xj = (Xj1, …, Xjd(j)), 1≤j≤D, are replaced by their corresponding DBN analogs For example, let us assume that some of the nodes (one or more) in Pa(Xi(t)) belong to the set of dependent nodes, say to the jth (1≤j≤D) dependent set. Then, those nodes are replaced by in The above parent sets are illustrated in Fig. 3, where, e.g., Pa(X i(t)) = {Xi(t −1), Xj1(t −1)} but because Xj1 is not independent.
Construction of the graph G1, then, goes as follows. For nodes 1≤i≤I, the graph has directed edges from time slice t − 1 to t such that the parents of are equal to For the grouped nodes 1≤j≤D, the graph has directed edges such that the parents of are
where , j1≤i≤jd(j), is an element of . This is illustrated in Fig. 3, where, e.g., Let us again expand the domain of predictor functions in F i by adding fictitious variables. However, the new domain of the functions is not assumed to be Pa(X j(t)) but, instead, it consists of the PBN counterparts of the nodes in An example is again shown in Fig. 3. For example, even though Pa(Xi(t)) = {X i(t − 1), Xj1 (t − 1)}, the PBN counterparts of are {X i(t − 1), Xj1 (t − 1), Xj2 (t − 1)}. It is easy to see that the local CPTs for the first I nodes can be formed as shown in Eq. (12). Let us then define D random vectors F j = (F(j1), …, F(jd(j))), 1≤j≤D, taking values in Fj1 × … × Fjd(j) and whose domains are expanded as discussed above. Thus, F j : 𝔹s → 𝔹jd(j), where s equals the number of nodes in the PBN counterparts of 5 Then, the CPT of the grouped node 1≤j≤D, can be computed as
where I(·) is the indicator function and the sum is expanded over all possible realizations of F j. The probability distribution of F j, in turn, can be computed by “integrating out” the other functions (see Eq. (1)).
4.2. A discrete-valued DBN as a dependent PBN
The final step is to show a way of expressing a discrete-valued DBN (B0, B1) as a PBN G(V, F). We again start by defining the nodes of a PBN. Discrete-valued nodes in the DBN are considered to have a binary representation. That is, let (a node in DBN) have b(i) bits in its binary representation in which case we can also write X̂i = (X̂i1,…, X̂ib(i)), X̂ij ∈ 𝔹, j = 1,…, b(i). For notational simplicity, we will assume in the following that the number of different values for each node is a power of two. In general, that does not need to be the case. For each node in the DBN, the corresponding PBN has a set of b(i) mutually dependent nodes. So, in total, the constructed PBN has nodes, where n equals the number of nodes in the given DBN.
The initial distribution of the PBN must be able to represent the same distribution as the first state of the given DBN. When the states of the DBN are considered via their binary representation it is clear, by the assumption stated in the beginning of Section 3, that the previous condition holds.
The joint probability distribution over the variables in the given discrete-valued DBN can be decomposed exactly the same way as shown in Eq. (9). Thus, the process is first order Markovian and we only need to consider the one step prediction probabilities, say from time t − 1 to time t. We cannot, however, define the predictor functions for each node in PBN independently. Fortunately, it suffices to consider the set of b(i) dependent nodes at a time.
Let us then construct the predictor functions for a PBN. Binary nodes in the DBN are not interesting since for those nodes we can use practically the same method as in Section 3.2. The only exception is that some parent nodes (in the DBN) may be non-binary. In that case, all the nodes in the PBN which correspond to a non-binary node in the DBN are used to predict the value of that gene. So, assume that we are considering a node which has b>1 bits in its binary representation. (The index i will be omitted from b since we concentrate on a single node at a time.) Let the corresponding nodes in the PBN be {Xi1, …, Xib}. The parents of the node are assumed to have q bits in total. Input variables of the predictor functions for the nodes {Xi1, …, Xib} are required to be the PBN counterparts of Further, all those predictor functions are mutually dependent.
Let the CPTs in Θ1 for the node be given: for all z ∈ 𝔹b, y ∈ 𝔹q. Assume that they are organized in 2q lists, each containing 2b entries (triplets): Ll = ((zl,1, y l, p l,1), …, (zl,2b, y l, p l,2b)) where zl,r ∈ 𝔹b y l ∈ 𝔹q and r = 1, …, 2b, l = 1, …, 2q. So, each list shows the elements of the CPT for a single parent (input) node configuration y l. Thus, the probabilities in each list must sum up to unity, i.e., , for all l = 1, …, 2q. For simplicity, let us assume that entries (zl,r, y l, p l,r) for which p l,r = 0 are removed from all lists. We now show a constructive algorithm that generates vector-valued random predictor functions F i : 𝔹q → 𝔹b for a set of dependent nodes in a PBN. Eventually, however, we will end up with a “normal” dependent PBN with standard Boolean functions for each node. In order to do so, we still introduce an additional set of 2q indices (pointers) . Index is used to index (point) an element in the ith list, and k is an iteration index. A constructive algorithm, for a single node in the DBN is shown in Fig. 4.
Fig. 4.

A constructive algorithm, for a single node in the DBN, for expressing a given DBN as a PBN.
In step 2, Eq. (17), we essentially define a function f k : 𝔹q → 𝔹b because yls are different for all lists l = 1, …, 2q. That is, the output value of f k is defined for all possible inputs. It is also important to note that in step 3, Eq. (18), we could have used as well because if , then . During each iteration we “subtract a probability mass” Pr{F i = f k} from all lists (distributions). So, after each iteration holds. This ensures that the condition in step 4 is meaningful and becomes valid such that all the sums , become equal to zero at the same time. Also, during each iteration, probability is set to zero and the index is incremented at least for one index l = 1, …, 2q. So, the criterion in step 4 is reached in at most after 2q × 2b iterations.
In order to see that the above procedure really constructs an equivalent PBN for the given DBN, consider one entry in the CPT of the given DBN, say . The above construction procedure generates a set of functions {f 1,…, f j}, with 2b≤j≤2q × 2b. It is easy to see that the sum of the selection probabilities of those functions for which f(y l) = zl,r is the probability . Thus, the same conditional probabilities are preserved. So, by applying the same construction method to all nodes in the DBN, one can see that all terms of Eq. (9) can be represented by the corresponding terms in Eq. (7).
To formalize the constructed PBN in the same manner as introduced in Section 2.1, one needs to define the predictor functions for each node separately. Given the vector-valued predictor functions {f 1, …, f j}, 2b≤j≤2q × 2b, then, for each node Xik, 1≤k≤b, one needs to “extract” all possible Boolean functions appearing as the kth element in the vector-valued functions. The probabilities of occurrence remain the same as set by the constructive method.
Thus, since any discrete-valued DBN can be represented as a dependent PBN, we can state the following theorem.
Theorem 3
Dependent PBNs G(V, F) and discrete-valued DBNs (B0, B1) whose initial and transition BNs B0 and B1 are assumed to have only within and between consecutive slice connections, respectively, can represent the same joint distribution over their corresponding variables.
The methods introduced in Sections 4.1 and 4.2 provide the actual conversions between the two modeling frameworks.
Example 4
We illustrate the operation of the above constructive procedure with a simple example. We apply it only to a single node, whose parents are . Thus, both nodes are quaternary, i.e., b = q = 2. Further, let the CPT for that node be given as in Table 4.
Table 4.
The local conditional probability distributions for the node
| z \ y | 00 | 01 | 10 | 11 |
|---|---|---|---|---|
| 00 | 0.1 | 0.2 | 0.1 | 0.2 |
| 01 | 0.5 | 0.2 | 0.3 | 0.4 |
| 10 | 0.2 | 0.3 | 0.5 | 0.3 |
| 11 | 0.2 | 0.3 | 0.1 | 0.1 |
Each entry in the table defines the probability that and, therefore, each column corresponds to a list Li, 1≤i≤4.
So, now each column in the table corresponds to a list Li, 1≤i≤4. In each list (column), the outputs, z, are in the same order for simplicity even though that does not need to be the case. So, when step 2 is applied for the first time, we define a function f 1 : 00↦00, 01↦00, 10↦00, 11↦00 with the selection probability Pr{F i = f 1} = 0.1. Note that, for each input (column), we pick the first non-zero element. Selected elements are shown in bold-face in Table 4 and the selection probability is the minimum of the selected elements. After step 3, the lists will be updated as shown in Table 5. During the second iteration one gets another vector-valued function (see Table 5) f 2 : 00↦01, 01↦00, 10↦01, 11↦00, and the corresponding selection probability is Pr{F i = f 2} = 0:1. The lists will again be updated as shown in Table 6. In a similar manner we obtain the third function f 3 : 00↦01, 01↦01, 10↦01, 11↦01, with the selection probability Pr{F i = f 1} = 0:2. The lists are updated as shown in Table 7. The remaining iterations are similar (not shown). Note that the sums of probabilities in each list (column) are the same within each iteration.
Table 5.
The lists Li, 1≤i≤4, after the first iteration of the algorithm
| z \ y | 00 | 01 | 10 | 11 |
|---|---|---|---|---|
| 00 | 0 | 0.1 | 0 | 0.1 |
| 01 | 0.5 | 0.2 | 0.3 | 0.4 |
| 10 | 0.2 | 0.3 | 0.5 | 0.3 |
| 11 | 0.2 | 0.3 | 0.1 | 0.1 |
Table 6.
The lists Li, 1≤i≤4, after the second iteration of the algorithm
| z \ y | 00 | 01 | 10 | 11 |
|---|---|---|---|---|
| 00 | 0 | 0 | 0 | 0 |
| 01 | 0.4 | 0.2 | 0.2 | 0.4 |
| 10 | 0.2 | 0.3 | 0.5 | 0.3 |
| 11 | 0.2 | 0.3 | 0.1 | 0.1 |
Table 7.
The lists Li, 1≤i≤4, after the third iteration of the algorithm
| z \ y | 00 | 01 | 10 | 11 |
|---|---|---|---|---|
| 00 | 0 | 0 | 0 | 0 |
| 01 | 0.2 | 0 | 0 | 0.2 |
| 10 | 0.2 | 0.3 | 0.5 | 0.3 |
| 11 | 0.2 | 0.3 | 0.1 | 0.1 |
The above procedure is not guaranteed to produce optimal sets of vector-valued functions. As in the case of independent networks, one can, e.g., minimize the number of functions. To set the stage, consider again the lists L1, 1≤l≤2q for a single node X i. The goal is to find the smallest set of vector-valued functions F i = {f 1, …, f m} together with their selection probabilities Pr{F i = f j} ∈ [0, 1] whose sum is equal to one, such that each pl,r can be written as
where Il,r ⊆ {1, …, m}, with the constraint that j ∈ Il,r ⇔ j ∉ Il,s, s ≠ r (j = 1, …, m and r = 1, …, 2b), for all l. The extra constraint essentially says that each individual function f j is deterministic, i.e, each input yl is mapped to a single output zl,r. Let J j = {(l, r)|j ∈ I l,r}, then the actual vector functions are defined as
and their validity can be checked as shown in Eq. (16). Efficient algorithms for the construction of the optimal vector-valued functions remain to developed.
5. Extensions of PBNs
The PBN model was further developed in [7] by introducing node perturbations. In the context of gene regulatory network modeling, the perturbations can capture various random or unknown factors, such as environmental conditions, that can possibly affect the expression value of a gene.
From a mathematical point of view, node perturbation can be defined in a variety of ways, but let us use the definition from [7]. At every time step of the network, we have a so-called perturbation vector γ ∈ 𝔹. The value of the ith node in the network is flipped if the corresponding element of γ is one. We will be assuming the perturbation vector to be independent and identically distributed (i.i.d.) for simplicity, even though this is not necessary in general. Let the probability of a single node perturbation be Pr{γi = 1} = p for all i, where γi denotes the ith element of γ. Let the random perturbation process also be homogeneous. Then, given the current state x(t − 1) of the network and a network realization f j, 1≤j≤N, the updating step of the network is
| (19) |
where ⊕ denotes addition modulo 2 and (1 − p)n is the probability of no perturbation occurring. That is, if no nodes are perturbed, the standard network transition function is used as explained in Section 2.1, while in the case of random node perturbations, the next state is determined by the previous state and the perturbation vector. An alternative definition would allow the unperturbed nodes to transition according to the (random) network function and only the perturbed nodes to be flipped.
Another extension to PBNs was introduced in [34] by defining an additional binary random variable δ which controls the random network changes. If δ = 1, then the network realization is randomly selected for the next time step as discussed in Section 2. Otherwise (δ = 0) the previously used network function is used. The network change variable is assumed to homogeneous with Pr{δ = 1} = q. With this extension the state of a PBN consists of the actual variables X as well as the network function F.
Effectively the same model can also be defined in the DBN context. Let us concentrate on independent PBNs for simplicity. The basis of the model is the same as illustrated in Section 3.1. The effects of perturbations and random network changes can be captured by adding additional hidden nodes γ(t), F(t) and δ(t), see Fig. 5.
Fig. 5.
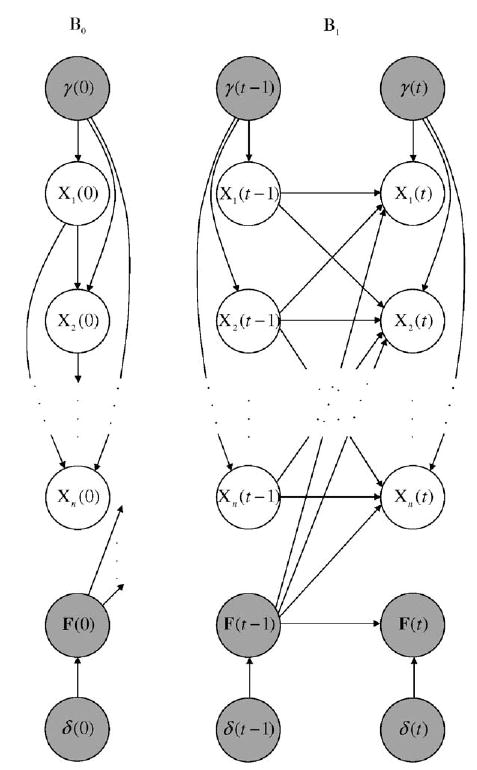
Basic building blocks of a dynamic Bayesian network (DBN) allowing node perturbations and random network changes. B0 and B1 are the initial and transition Bayesian networks (BNs). The hidden variables γ(t), F(t) and δ(t) are shown in gray.
Concerning the node perturbations, the hidden variable γ(t) is assumed to have n-dimensional independent Bernoulli distribution with common probability p. The perturbation node γ(t) is also added into the set of parent nodes of i.e., for all i and t ≥ 0. The local CPTs of the nodes are the same as in Section 3.1, except with the following adaptation: for the parent node configurations where γ(t) = (00 … 0) the CPTs remain unchanged and only depend on the value of and for the remaining parent node configurations (where γ(t) ≠ (00 … 0), regardless of the value of the value of the node is xi(t) = xi(t − 1) ⊕ γi(t) with probability one. Also note that must belong to for all 1≤i≤n.
The random network changes can be accounted for using the additional hidden variables F(t) and δ(t). The network change variable δ(t) is Bernoulli with parameter q. The value of the network function F(t) depends directly on the value of F(t − 1) and δ(t), i.e., Pa(F(t)) = {F(t − 1), δ(t)}. If δ(t) = 0, then f(t) = f(t − 1). Otherwise a new network function is selected according to its selection probability. In the DBN formalism, the parents of the actual state variable, include not only its parent variables as defined in Section 3.1 but also the hidden network function variable, i.e., Given the value of the parent variables, the CPTs of the actual state variables are degenerate since each network function operates deterministically. In the case of random network changes, the effects of node perturbations can be handled as explained above.
6. Benefits of relationships between PBNs and DBNs
As already discussed in Section 1, having shown the connection between PBNs and DBNs (Theorems 1 and 3), it immediately opens up the possibility of applying the advanced tools of PBNs to DBNs and vice versa. Let us give a more detailed description of the tools that become available for DBNs and PBNs in Sections 6.1 and 6.2, respectively.
6.1. Benefits for DBNs
Relationships between PBNs and DBNs make the following tools, among others, available in the context of DBNs as well. The tools originally developed for PBNs can be applied in the context DBNs, e.g., by using the detailed conversion of a DBN to a PBN. Alternatively, knowing the detailed mapping of a DBN to a PBN and that the two models can capture the same probabilistic behavior, it is possible to tailor each PBN method to be used directly in the DBN framework. The details of these tailored methods are not provided here.
In the context of genetic regulatory networks, one may want to elicit certain long-run behavior from the network. For example, a certain set of states can be deemed “undesirable” and one may wish for the network to transition into a “desirable” set of states. For example, in terms of cancer therapeutics, one may want to reach the set of states representing apoptosis in order to suppress the growth of cancer cells, which may keep proliferating.
The problem of controlling the stationary behavior of the dynamic network model has recently been addressed in several papers. Shmulevich et al. [7] considered the control by means of gene interventions. As the intervention is typically only transient, it does not affect the steady-state distribution. Thus, one may want to achieve the desired behavior as quickly as possible. For computational reasons, the control should also be achieved by intervening with as few genes as possible. A control method using the first-passage times of Markov chains was used in [7]. A related approach was taken in [8] where the control was formulated in terms of (permanently) modifying the network structure. That is, given the set of undesirable and desirable states and the original stationary distribution of the network, the goal is to find the best alteration of some preselected number of Boolean predictor functions such that a desired effect in the resulting stationary distribution is obtained. Yet another approach to control the stationary behavior, under the assumption of external variables, was introduced in [9]. Datta et al. based their approach on the theory of controlled Markov chains and used dynamic programming to find the optimal sequence of control actions that minimize a performance index over a finite time horizon. To our knowledge, no methods for controlling the stationary behavior of DBNs have been introduced so far. Therefore, the aforementioned methods can be of great value for the applications of DBNs.
Owing to the large number of variables in full genetic regulatory networks, it is often necessary to constrain one’s approach to sufficiently small sub-networks. Therefore, network projections onto sub-networks are of particular interest. Dougherty and Shmulevich considered mappings between PBNs, such as projections, node adjunctions and resolution reductions, while at the same time preserving consistency with the original probabilistic structure [10].
Learning of PBN models has been considered e.g. in [11–13,15]. A promising approach to network learning was taken in [12]. In order to limit the search space, a novel clustering-based approach to finding sets of possible predictor genes for each gene was used. Further, a reversible jump Markov chain Monte Carlo method together with the coefficient of determination were used to compute the model order and parameters. Computationally more efficient learning schemes, although with the cost of decreased accuracy, were studied in [13]. From the DBN point of view, this can be viewed as an approximate learning method.
It is also worth noting that the constructed predictor functions can provide more compact presentation of the CPTs in DBNs. For example, instead of storing the whole CPT consisting of 2q+1 real numbers one can represent the conditional probabilities using PBNs. Assuming only m predictor functions are required to represent a CPT, then only m 2q-length binary vectors (assuming truth table format for the Boolean functions) together with m real number (selection probabilities) are needed.
6.2. Benefits for PBNs
Having shown the relationships between PBNs and DBNs (Theorems 1 and 3), the following tools from the context of DBNs can be used for PBNs as well. As introduced in the benefits for DBNs section, the DBN tools can be applied to PBNs by using the detailed mapping of a PBN to a DBN. Alternatively, the methods originally developed for DBNs can be tailored, with the help of the detailed conversions between the two model classes, to be used directly in the PBN context.
Gene expression measurements are often corrupted by missing data values. If it so happens that the missing components are of particular relevance, one may want to compute the posterior probability of the hidden nodes, given the incomplete measurements. In order to compute that, one can use exact inference methods [35] in the context of BNs, or approximate inference methods [36] if the exact solution is intractable.
Learning of gene regulatory networks from data has turned out to be a difficult problem and, therefore, efficient and flexible tools are needed. From a theoretical point of view, optimal learning of graphical models from data is a difficult problem—the optimal learning of BN structure using a Bayesian metric, BDe score, was shown to be an NP-complete problem [37], and the temporal dimension of DBNs does not make the problem any easier. In principle, learning of graphical models can be divided into two groups, model selection and parameter estimation, the former step generally being considerably harder than the latter. As is apparent based on the relationships between PBNs and DBNs, the same division can also be used when learning PBNs. Now, the model selection step corresponds to the selection of variables for each Boolean predictor function, whereas the parameter estimation step is related to the selection of the optimal Boolean predictor functions along with their selection probabilities.
Learning of DBNs has been a topic of several studies. A good introduction to learning of (non-temporal) BNs can be found in [38,39]. Learning of DBNs has been studied, e.g., in [17,40]. More efficient inference methods based on estimating local properties (Markov blanket) of the underlying graph have also been introduced [41,42].
In the fully observable case, learning the network structure should not be overly complicated at least for small indegree, which is the maximum number of parents of each node. However, computational and theoretical problems may arise in the case of incomplete data, which is quite common in the context of gene expression measurements. Also, unknown control/regulatory factors not taken into account in the network model can be considered as hidden variables. The expectation-maximization (EM) algorithm is a standard tool for tackling the problems caused by missing data [17,43]. A more efficient structural EM (SEM) algorithm has been introduced for learning BNs (see, for instance, [17]). However, as the EM is an optimization routine, computational costs increase and there is no guarantee for the global maxima.
Data used in the gene regulatory network learning are usually both expensive and difficult to collect. Therefore, one should carefully design the experiments in order to gain maximal advantage. The use of active learning can remarkably reduce the number of observations required to learn the model by choosing data instances whose expected decrease in the uncertainty in the model learned so far is the greatest. Greedy active learning methods for BNs have recently been introduced in [44–46].
As a summary, a number of advanced methods have been developed for learning DBNs. Since PBNs and DBNs can capture the same probabilistic behavior, a straightforward way of using the DBN learning methods in the context of PBNs is as follows: learn a DBN from a given data set, e.g. by maximizing the posterior probability, and then convert it to a PBN using the methods introduced above. However, special care must be taken in the conversion of a DBN to a PBN due to the one-to-many nature (non-uniqueness) of this transformation.
7. Discussion
The subclass of DBNs used in this study, even though not necessarily binary-valued, is by far the most commonly used DBN model class in the context of modeling dynamic gene regulatory networks. In particular, often only between-slice connections are used since that fits well in the modeling of cause-effect behavior. However, independent PBNs cannot cope with dependent effects (or nearly instantaneous interactions) between variables. The same applies to the binary-valued DBNs as well. This issue can be handled using dependent PBNs or discrete-valued (i.e., binary vector-valued) DBNs. Indeed, the discrete-valued DBNs can be viewed as binary DBNs where some individual binary variables (within slice) are clustered together, hence forming binary vector-valued variables. Since the binary vector-valued nodes can have any local conditional distributions, the individual elements of a binary vector variable can have any joint effects. The same applies to dependent PBNs due to the correspondence between the models.
In the context of learning genetic regulatory networks, the main focus so far has been on the use of gene expression data only. In the context of PBNs, several papers have been published on learning the network structure e.g. [12–15] most of which use the so-called coefficient of determination (CoD) principle [47]. Learning methods for DBNs, in the context of genetic networks, have been studied in [17,21,22,24,25,27,29]. Note, however, that some of the DBN studies have concentrated on non-temporal BNs. A problem of non-dynamic model inference is that one generally loses the causal direction of regulatory effects. This can be circumvented by using perturbations, such as gene over-expression and knock-outs [28,30].
As the DBN is a versatile model class, different information sources can be used, in a principled way, in the model inference. Recently, the use of so-called location data [48], measuring the protein–DNA interactions, was introduced in the framework of BNs [23], and with more abundant data in [49].6 The optimal DBN model structures are the ones that maximize the a posteriori probability of the model ℳ given the data 𝒟, Pr{ℳ|𝒟} ∝ Pr{𝒟| ℳ} Pr{ℳ}. In [23], the location data was brought into the model inference via the model prior Pr{ℳ}. Loosely speaking, genome-wide location data can be used to measure the degree to which a transcriptional factor, a product of a gene or several genes, binds to the promoter of a gene. So, that data provides a measure of plausibility of a certain gene being a direct parent of some other (or even the same) gene. In other words, by using location data we may be able to assess the likelihood of X j(t − 1) ∈ Pa(X i(t)).
In order to incorporate even more prior knowledge into the network learning process, we may also consider the use of sequence analysis results, possibly combined with clustering results. For instance, genes having highly homologous promoter sequences are likely to be regulated by the same transcriptional factors. An approach combining both expression data and sequence information for finding putative regulatory elements was introduced in [50] (for further reading see e.g. [51] and references therein). A purely combinatorial approach to the same problem, even though it was also validated by expression data, was introduced in [52]. Assume that two genes, say Xi and Xj, are found to have highly homologous promoter sequences and are hypothesized to be regulated by the same transcriptional factor, say a product of gene Xk. Then, one can expect that Xk(t − 1) ∈ Pa(Xi(t)) ⇔ Xk(t − 1) ∈ Pa(Xj(t)), where the “if and only if” must be considered, of course, probabilistically, meaning that the probability of this condition can be expected to be high.
In other words, basically any additional information source can be utilized in the process of learning gene regulatory network structure (for a recent study, see [26]). For instance, recent preliminary results indicate that the genetic regulatory networks exhibit scale-free topology, as do many real-world networks [53,54]. All aforementioned information sources can be included in the model induction process through the model prior. Model inference, which utilizes several different information sources, is a topic for further studies.
Another interesting implication of the proposed relationships between the models can be seen after rewriting Pr{ℳ|𝒟} as Pr{ℳ|𝒟} ∝ (∫Pr{𝒟| ℳ, θ} Pr{θ| ℳ}) Pr{ℳ}, where θ denotes the model parameters. Because the model parameters in DBNs correspond to the predictor functions and their selection probabilities in PBNs, one can also use the natural constraints of the predictor function classes in the learning phase in a Bayesian fashion. For instance, Harris et al. [55] recently studied more than 150 known regulatory transcriptional systems with varying number of regulating components and found that these controlling rules are strongly biased toward so-called canalizing functions. These and other natural constraints on the class of rules in genetic regulatory networks [56] can be incorporated in the learning phase.
In summary, we have shown relationships between dynamic Bayesian networks and probabilistic Boolean networks. These relationships between the models extend the collection of advanced analysis tools for both model classes. Further, as the theory of both DBNs and PBNs is under vigorous research, new advances are directly applicable to the gene regulatory networks under both models.
Acknowledgments
This study was supported by Tampere Graduate School in Information Science and Engineering (TISE) (H. L.), Academy of Finland (H. L. and O. Y.-H.) and NIH R01 GM072855-01 (I. S.).
Footnotes
The dynamics of PBNs can be studied using Markov chains.
Throughout this paper, symbol X is used for both (random) vector variables and sets of (random) variables so that notations such as Xj ∩ Xk make sense.
In order to follow the standard notation we write Pr{x} instead of Pr{X = x}.
Note that is essentially the unity function. Also, some of the Boolean expressions in Eq. (13) can possibly be expressed in more compact form.
For example, in Fig. 3, the PBN counterparts of are {Xi(t −1), Xj1 (t −1), Xj2 (t −1), Xk1 (t −1), Xk2 (t −1)}.
Bayesian networks were not used in [49].
References
- 1.de Jong H. Modeling and simulation of genetic regulatory systems: a literature review. J Comput Biol. 2002;9(1):67–103. doi: 10.1089/10665270252833208. [DOI] [PubMed] [Google Scholar]
- 2.de Hoon MJL, Imoto S, Kobayashi K, Ogasawara N, Miyano S. Inferring gene regulatory networks from time-ordered gene expression data of Bacillus subtilis using differential equations. Proceedings of the Pacific Symposium on Biocomputing. 2003;8:17–28. [PubMed] [Google Scholar]
- 3.Sakamoto E, Iba H. Inferring a system of differential equations for a gene regulatory network by using genetic programming. Proceedings of the Congress on Evolutionary Computation ’01. 2001:720–726. [Google Scholar]
- 4.Shmulevich I, Dougherty ER, Seungchan K, Zhang W. Probabilistic Boolean networks: a rule-based uncertainty model for gene regulatory networks. Bioinformatics. 2002;18(2):261–274. doi: 10.1093/bioinformatics/18.2.261. [DOI] [PubMed] [Google Scholar]
- 5.Kauffman SA. Metabolic stability and epigenesis in randomly constructed genetic nets. J Theoret Biol. 1969;22(3):437–467. doi: 10.1016/0022-5193(69)90015-0. [DOI] [PubMed] [Google Scholar]
- 6.Kauffman SA. The Origins of Order: Self-organization and Selection in Evolution. Oxford University Press; New York: 1993. [Google Scholar]
- 7.Shmulevich I, Dougherty ER, Zhang W. Gene perturbation and intervention in probabilistic Boolean networks. Bioinformatics. 2002;18(10):1319–1331. doi: 10.1093/bioinformatics/18.10.1319. [DOI] [PubMed] [Google Scholar]
- 8.Shmulevich I, Dougherty ER, Zhang W. Control of stationary behavior in probabilistic Boolean networks by means of structural intervention. J Biol Systems. 2002;10(4):431–445. [Google Scholar]
- 9.Datta A, Choudhary A, Bittner ML, Dougherty ER. External control in Markovian genetic regulatory networks. Mach Learning. 2003;52(1–2):169–181. doi: 10.1093/bioinformatics/bth008. [DOI] [PubMed] [Google Scholar]
- 10.Dougherty ER, Shmulevich I. Mappings between probabilistic Boolean networks. Signal Processing. 2003;83(4):745–761. doi: 10.1016/j.sigpro.2005.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lähdesmäki H, Shmulevich I, Yli-Harja O. On learning gene regulatory networks under the Boolean network model. Mach Learning. 2003;52(1–2):147–167. [Google Scholar]
- 12.Zhou X, Wang X, Dougherty ER. Construction of genomic networks using mutual-information clustering and reversible-jump Markov-chain-Monte-Carlo predictor design. Signal Processing. 2003;83(4):745–761. [Google Scholar]
- 13.Hashimoto RF, Dougherty ER, Brun M, Zhou ZZ, Bittner ML, Trent JM. Efficient selection of feature sets possessing high coefficients of determination based on incremental determinations. Signal Processing. 2003;83(4):695–712. [Google Scholar]
- 14.Kim S, Dougherty ER, Bittner ML, Chen Y, Sivakumar K, Meltzer P, Trent JM. General nonlinear framework for the analysis of gene interaction via multivariate expression arrays. J Biomedical Optics. 2000;5(4):411–424. doi: 10.1117/1.1289142. [DOI] [PubMed] [Google Scholar]
- 15.Kim S, Li H, Dougherty ER, Cao N, Chen Y, Bittner M, Suh EB. Can Markov chain models mimic biological regulation? J Biol Systems. 2002;10(4):431–445. [Google Scholar]
- 16.Dean T, Kanazawa K. A model for reasoning about persistence and causation. Comput Intell. 1989;5(3):142–150. [Google Scholar]
- 17.Friedman N, Murphy K, Russell S. Learning the structure of dynamic probabilistic networks, Proceedings of the Fourteenth Conference on Uncertainty in Artificial Intelligence (UAI); 1998. pp. 139–147. [Google Scholar]
- 18.Murphy K. Dynamic Bayesian Networks: Representation, Inference and Learning. University of California, Berkeley, Computer Science Division; July 2002. [Google Scholar]
- 19.Zweig G, Russell S. Madison, Wisconsin: 1998. Speech recognition with dynamic Bayesian networks, in: Proceedings of the Fifteenth National Conference on Artificial Intelligence AAAI-98; pp. 173–180. [Google Scholar]
- 20.Chang K, Fung R. Target identification with Bayesian networks in a multiple hypothesis tracking system. Optical Eng. 1994;36(3):684–691. [Google Scholar]
- 21.Friedman N, Linial M, Nachman I, Pe’er D. Using Bayesian networks to analyze expression data. J Comput Biol. 2000;7:601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- 22.Hartemink A, Gifford D, Jaakkola T, Young R. Using graphical models and genomic expression data to statistically validate models of genetic regulatory networks. Proceedings of the Pacific Symposium on Biocomputing. 2001;6:422–433. doi: 10.1142/9789814447362_0042. [DOI] [PubMed] [Google Scholar]
- 23.Hartemink A, Gifford D, Jaakkola T, Young R. Combining location and expression data for principled discovery of genetic regulatory network models. Proceedings of the Pacific Symposium on Biocomputing. 2002;7:437–449. [PubMed] [Google Scholar]
- 24.Imoto S, Goto T, Miyano S. Estimation of genetic networks and functional structures between genes by using Bayesian networks and nonparametric regression. Proceedings of the Pacific Symposium on Biocomputing. 2002;7:175–186. [PubMed] [Google Scholar]
- 25.Imoto S, Kim S, Goto T, Aburatani S, Tashiro K, Kuhara S, Miyano S. Bayesian network and nonparametric heteroscedastic regression for nonlinear modeling of genetic network. J Bioinformatics Comput Biol. 2003;1(2):231–252. doi: 10.1142/s0219720003000071. [DOI] [PubMed] [Google Scholar]
- 26.Imoto S, Higuchi T, Goto T, Tashiro K, Kuhara S, Miyano S. Combining microarrays and biological knowledge for estimating gene networks via Bayesian networks. Proceedings of the Second Computational Systems Bioinformatics (CSB’03) 2003:104–113. [PubMed] [Google Scholar]
- 27.K. Murphy, S. Mian, Modelling gene expression data using dynamic Bayesian networks, Technical Report, University of California, Berkeley, 1999.
- 28.Pe’er D, Regev A, Elidan G, Friedman N. Inferring subnetworks from perturbed expression profiles. Bioinformatics. 2001;17(Suppl 1):215S–224S. doi: 10.1093/bioinformatics/17.suppl_1.s215. [DOI] [PubMed] [Google Scholar]
- 29.Perrin B-E, Ralaivola L, Mazurie A, Bottani S, Mallet J, d’Alché-Buc F. Gene networks inference using dynamic Bayesian networks. Bioinformatics. 2003;19(Suppl 2):ii138–ii148. doi: 10.1093/bioinformatics/btg1071. [DOI] [PubMed] [Google Scholar]
- 30.Yoo C, Thorsson V, Cooper GF. Discovery of causal relationships in a gene-regulation pathway from a mixture of experimental and observational DNA microarray data. Proceedings of the Pacific Symposium on Biocomputing. 2002;7:498–509. doi: 10.1142/9789812799623_0046. [DOI] [PubMed] [Google Scholar]
- 31.Pearl J. Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Interference. Morgan Kaufmann Publishers Inc.; Los Altos, CA: 1988. [Google Scholar]
- 32.Neapolitan R. Probabilistic Reasoning in Expert Systems. Wiley; New York: 1990. [Google Scholar]
- 33.Wegener I. The Complexity of Boolean Functions. Wiley; New York: 1987. [Google Scholar]
- 34.Zhou X, Wang X, Pal R, Ivanov I, Bittner ML, Dougherty ER. A Bayesian connectivity-based approach to constructing probabilistic gene regulatory networks. Bioinformatics. 2004;20(17):2918–2927. doi: 10.1093/bioinformatics/bth318. [DOI] [PubMed] [Google Scholar]
- 35.Huang C, Darwiche A. Inference in belief networks: a procedural guide. Internat J Approx Reason. 1996;15(3):225–263. [Google Scholar]
- 36.Jordan MI, Ghahramani Z, Jaakkola T, Saul LK. An introduction to variational methods for graphical models. Mach Learning. 1999;37(2):183–233. [Google Scholar]
- 37.Chickering DM. Learning Bayesian networks is NP-complete. In: Fisher D, Lenz H-J, editors. Learning from Data: Artificial Intelligence and Statistics V. Springer; Berlin: 1996. pp. 121–130. [Google Scholar]
- 38.D. Heckerman, A tutorial on learning with Bayesian networks, Technical Report MSR-TR-95-06, Microsoft Corporation, Redmond, USA, 1996.
- 39.Pearl J. Learning Bayesian Networks. Prentice-Hall; Englewood Cliffs, NJ: 2003. [Google Scholar]
- 40.Ghahramani Z. Learning dynamic Bayesian networks. In: Giles CL, Gori M, editors. Adaptive Processing of Sequences and Data Structures, Lecture Notes in Artificial Intelligence. Springer; Berlin: 1998. pp. 168–197. [Google Scholar]
- 41.Hwang KB, Lee JW, Chung SW, Zhang B-T. Lecture Notes in Artificial Intelligence. Vol. 2417. 2002. Construction of large-scale Bayesian networks by local to global search; pp. 375–384. [Google Scholar]
- 42.Margaritis D, Thrun S. Bayesian network induction via local neighborhoods. Proceedings of the Advances in Neural Information Processing Systems. 2000;12:505–511. [Google Scholar]
- 43.Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J Roy Stat Soc Ser B. 1977;39:1–38. [Google Scholar]
- 44.K. Murphy, Active learning of causal Bayes net structure, Technical Report, University of California, Berkeley, March 2001.
- 45.Tong S, Koller D. Active learning for parameter estimation in Bayesian networks; Proceedings of the Neural Information Processing Systems; 2000. pp. 647–653. [Google Scholar]
- 46.Tong S, Koller D. Active learning for structure in Bayesian networks; Proceedings of the International Joint Conference on Artificial Intelligence; 2001. pp. 863–869. [Google Scholar]
- 47.Dougherty ER, Kim S, Chen Y. Coefficient of determination in nonlinear signal processing. Signal Processing. 2000;80(10):2219–2235. [Google Scholar]
- 48.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290(5500):2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 49.Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298(5594):799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 50.Vilo J, Brazma A, Jonassen I, Robinson A, Ukkonen E. Mining for putative regulatory elements in the yeast genome using gene expression data; Proceedings of the International Conference on Intelligent Systems for Molecular Biology; pp. 384–394. [PubMed] [Google Scholar]
- 51.Baldi P, Hatfield GW. DNA Microarrays and Gene Expression: From Experiments to Data Analysis and Modeling. Cambridge University Press; Cambridge, MA: 2002. [Google Scholar]
- 52.Pilpel Y, Sudarsanam P, Church GM. Identifying regulatory networks by combinatorial analysis of promoter elements. Nature Genetics. 2001;29(2):153–159. doi: 10.1038/ng724. [DOI] [PubMed] [Google Scholar]
- 53.Fox JJ, Hill CC. From topology to dynamics in biochemical networks. Chaos. 2001;11(4):809–815. doi: 10.1063/1.1414882. [DOI] [PubMed] [Google Scholar]
- 54.Oosawa C, Savageau MA. Effects of alternative connectivity on behavior of randomly constructed Boolean networks. Physica D. 2002;170(2):143–161. [Google Scholar]
- 55.Harris SE, Sawhill BK, Wuensche A, Kauffman S. A model of transcriptional regulatory networks based on biases in the observed regulation rules. Complexity. 2002;7(4):23–40. [Google Scholar]
- 56.Shmulevich I, Lähdesmäki H, Dougherty ER, Astola J, Zhang W. The role of certain post classes in Boolean network models of genetic networks. Proc Natl Acad Sci USA. 2003;100(19):10734–10739. doi: 10.1073/pnas.1534782100. [DOI] [PMC free article] [PubMed] [Google Scholar]