

Abstract
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant hereditary disorder characterized by multiple parathyroid, pancreatic, duodenal, and pituitary neuroendocrine tumors. Nonendocrine mesenchymal tumors, such as lipomas, collagenomas, and angiofibromas have also been reported. MEN1-associated neuroendocrine and some mesenchymal tumors have documented MEN1 gene alterations on chromosome 11q13. To test whether the MEN1 gene is involved in the pathogenesis of multiple smooth muscle tumors, we examined the 11q13 loss of heterozygosity (LOH) and clonality patterns in 15 leiomyomata of the esophagus, lung, and uterus from five patients with MEN1. Forty sporadic uterine leiomyomata were also studied for 11q13 LOH. LOH analysis was performed using four polymorphic DNA markers at the MEN1 gene locus; D11S480, PYGM, D11S449, and INT-2. 11q13 LOH was detected in 10 of 12 (83%) MEN1-associated esophageal and uterine smooth muscle tumors. In contrast, LOH at the MEN1 gene locus was demonstrated only in 2 of 40 (5%) sporadic uterine tumors. LOH at 11q13 was not documented in three lung smooth muscle tumors from a single patient with MEN1. Ten tumors from two female patients were additionally assessed for clonality by X-chromosome inactivation analysis. The results demonstrated different clonality patterns in multiple tumors in the same organ in each individual patient. The data indicate that leiomyomata of the esophagus and uterus in MEN1 patients arise as independent clones, develop through MEN1 gene alterations, and are an integral part of MEN1. However, the MEN1 gene is not a significant contributor to the tumorigenesis of sporadic uterine leiomyomata.
The gene for multiple endocrine neoplasia type 1 (MEN1), an autosomal dominant tumor syndrome, has been mapped to chromosome 11q131 and recently identified.2 The MEN1 gene is thought to act as a tumor suppressor based on the presence of inherited inactivating mutations in the constitutional DNA of affected family members accompanied by the loss of the wild-type allele in associated tumors.2-4 Somatic inactivation of the MEN1 gene has been also documented in a subset of sporadic counterpart parathyroid, enteropancreatic, and pulmonary endocrine tumors, and mesenchymal tumors.5
MEN1 patients typically present first with primary hyperparathyroidism resulting from multiple parathyroid tumors caused by MEN1 gene alterations.6 Neuroendocrine tumors of the pancreas, duodenum, anterior pituitary gland, stomach, and lung are other tumors that are an integral part of MEN1.7,8 Nonendocrine mesenchymal tumors, such as lipomas, angiofibromas, and collagenomas, have also been shown to be associated with MEN1 and MEN1 gene alterations.8-10 Leiomyomata have been occasionally documented in MEN1 patients,8,11-14 and loss of heterozygosity (LOH) at the MEN1 locus was recently shown in two esophageal leiomyomata from one MEN1 patient.15 MEN1 gene inactivation in lung or uterine leiomyomata, however, has not been studied. To test whether MEN1 gene alterations are involved in the development of multiple smooth muscle tumors in MEN1 patients, we analyzed 15 leiomyomata from five patients with documented MEN1 germline mutations for LOH at the MEN1 gene locus. To assess whether MEN1 gene alterations are present in sporadic smooth muscle tumors, we analyzed 40 sporadic uterine leiomyomata for MEN1 gene deletion for comparison. Furthermore, to evaluate whether MEN1-associated leiomyomata arise as independent clonal events at different anatomical sites within an organ, the patterns of 11q13 LOH in different tumors from individual patients were compared, and X-chromosome inactivation analysis was performed.
Materials and Methods
Patients and Tumors
Five MEN1 patients who had leiomyomata were enrolled in a protocol approved by the Institutional Review Board of the National Institute of Diabetes, Digestive, and Kidney Diseases and gave informed consent. Each patient met clinical criteria for MEN16 and had a MEN1 germline mutation16 (Table 1)▶ . Endocrine and mesenchymal MEN1 tumor manifestations included multiple parathyroid tumors, gastrinomas and metastases, pancreatic neuroendocrine tumors, lung carcinoids, lipomas, and angiofibromas. The tumor expressions in the five patients were classical of MEN1, as were the expressions in the other affected members of their families (data not shown). Each patient had a different germline MEN1 mutation. Like typical MEN1 mutations, they were distributed across the open reading frame and four predicted a menin truncation whereas one (kdel119) predicted a missense change (Table 1)▶ . There was neither a specific phenotype nor a specific genotype associated with leiomyomata in the five cases. Fifteen formalin-fixed, paraffin-embedded leiomyomata that were available and yielded DNA suitable for analysis were included in the study.
Table 1.
Clinical and Germline Mutation Data in Five MEN1 Patients with 15 Smooth Muscle Tumors
| Patient no. | Age MEN1*/age leiomyoma† | Sex | Leiomyoma site (no. of tumors‡) | MEN1 germline mutation | MEN1-associated tumors |
|---|---|---|---|---|---|
| 1 | 36 /57 | F | Esophagus (1) | kdel119 | Parathyroid tumors |
| Gastrinomas | |||||
| Lipoma | |||||
| 2 | 29 /51 | F | Esophagus (2) | 512delC | Parathyroid tumors |
| Gastrinomas | |||||
| Pancreatic neuroendocrine tumors (nonfunctioning) | |||||
| Lung carcinoid | |||||
| 3 | 35 /56 | F | Uterus (3) | 1650insC | Parathyroid tumors |
| Gastrinomas | |||||
| Pancreatic neuroendocrine tumors (nonfunctioning) | |||||
| 4 | 34 /40 | F | Lung (3) | 735del4 | Parathyroid tumors |
| Esophagus (1) | |||||
| Uterus (3) | |||||
| 5 | 36 /35 | F | Esophagus (2) | 11202del2 | Parathyroid tumors |
| Gastrinomas | |||||
| Metastatic gastrinoma | |||||
| Pancreatic neuroendocrine tumors (nonfunctioning) | |||||
| Lipoma | |||||
| Angiofibromas |
*Patient’s age at time of clinical diagnosis of MEN1.
†Patient’s age at time of leiomyoma diagnosis.
‡Number of smooth muscle tumors available for LOH analysis.
Forty sporadic uterine leiomyomata were obtained from archival tissue blocks. Tumors were selected only when the patient’s medical history was available and revealed no evidence of clinical signs or family history of MEN1 including parathyroid tumors, pituitary tumors, and enteropancreatic tumors.
11q13 LOH Analysis
Briefly, 6-μm serial sections of each tumor were stained with hematoxylin and eosin, and evaluated for verification of leiomyoma diagnosis. An adjacent tissue section was used for DNA procurement. Tumor tissue was selectively microdissected from normal tissue as previously described.17 Two to five leiomyomatous areas were microdissected from each section under light microscopic guidance and placed in proteinase K buffer for DNA extraction. Patient-matched DNA from blood or normal tissue, and neuroendocrine tumors were used for comparison where appropriate with normal and neuroendocrine tumor tissue. DNA was amplified by polymerase chain reaction with microsatellite markers; D11S480, PYGM (CAGA or AT), D11S449, and INT-2 (Table 2)▶ .18-20 The markers are listed from centromeric (left) to telomeric (right), with the MEN1 gene bounded by PYGM and D11S449.2 The amplification products were visualized by polyacrylamide gel electrophoresis and autoradiography. The case was considered to be informative for a polymorphic marker on 11q13 if normal tissue DNA showed two different alleles (heterozygosity). A reduction of the intensity of one allele in tumor DNA of 70% or greater, verified by phosphoimage intensity analysis as indicated, was interpreted as LOH (Figure 1)▶ . All patients were informative for at least two tested markers at 11q13.
Table 2.
Results of 11q13 LOH and X-Chromosome Inactivation Analysis in 15 Leiomyomata from Five Patients with MEN1
| Patient no. | Tumor site | Tumor no. | D11S480* | PYGM† | D11S449 | INT-2 | HUMARA‡ |
|---|---|---|---|---|---|---|---|
| 1 | Esophagus | T1 | • | NI | NI | • | — |
| 2 | Esophagus | T2 | • | • | • | • | — |
| Esophagus | T3 | • | • | • | • | — | |
| 3 | Uterus | T4 | • | NI | • | NI | Monoclonal(U) |
| Uterus | T5 | • | NI | • | NI | Monoclonal(L) | |
| Uterus | T6 | NI | NI | Monoclonal(L) | |||
| 4 | Lung | T7 | NI | NI | Monoclonal(U) | ||
| Lung | T8 | NI | NI | Monoclonal(U) | |||
| Lung | T9 | NI | NI | Monoclonal(U) | |||
| Esophagus | T10 | Ni | NI | Polyclonal (U, L) | |||
| Uterus | T11 | NI | • | • | NI | Monoclonal(U) | |
| Uterus | T12 | NI | • | NI | Monoclonal(L) | ||
| Uterus | T13 | NI | • | • | NI | Monoclonal(L) | |
| 5§ | Esophagus | T14 | NI | • | • | NI | — |
| Esophagus | T15 | NI | • | • | NI | — |
•, LOH; , retention of heterozygosity; NI, noninformative; —, not done.
*Chromosome 11q13 polymorphic markers at the MEN1 locus are listed in order from centromeric (left) to telomeric (right), MEN1 is between PYGM and D11S449.
†Marker PYGM(CAGA) in Patients 1 and 3, and marker PYGM(AT) in patients 4 and 5.
‡Monoclonal(U), only upper allele methylation in tumor as compared to normal tissue; monoclonal(L), only lower allele methylation in tumor as compared to normal tissue; polyclonal(U,L), both upper and lower alleles methylated in tumor.
§LOH data in two esophageal leiomyomata in patient 5 were previously reported,14 and reanalyzed and confirmed in this study.
Figure 1.

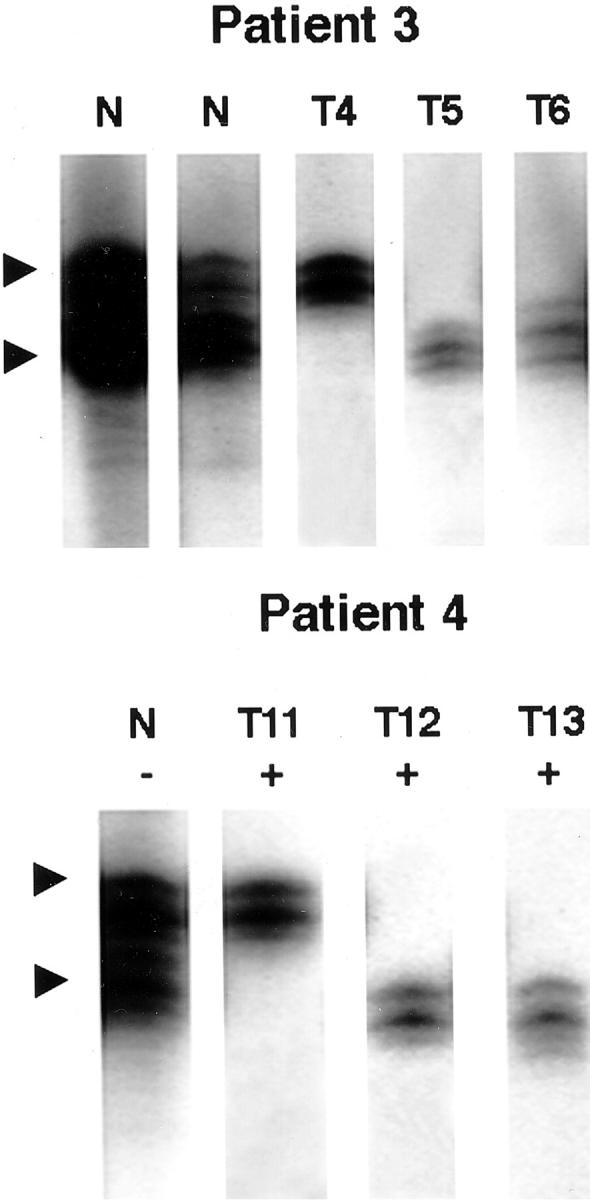
Representative results of 11q13 LOH in multiple leiomyomata in four MEN1 patients. Polymorphic markers (D11S480, PYGM, D11S449, INT-2) at the MEN1 gene locus. Tumor number corresponds to the tumor number in Table 2▶ : T1, esophageal tumor in patient 1; T2 and T3, esophageal tumors in patient 2; T4 to T6, uterine tumors in patient 3; T7 to T9, lung tumors in patient 4; T10, esophageal tumor in patient 4; T11 to T13, uterine tumors in patient 4. Arrowhead indicates the position of the two alleles. Deletion of the lower allele with marker D11S480 was detected in tumor T1 in patient 1 and in tumors T2 and T3 in patient 2. Deletion of the upper allele with marker D11S480 was detected in tumors T4 and T5 in patient 3. For marker PYGM, deletion of the lower allele was seen in tumors T2 and T3 in patient 2 (PYGM(CAGA)) and T11 to T13 in patient 4 (PYGM(AT)). Tumors T7, T8, T10 in patient 4 showed retention of heterozygosity with PYGM. For marker D11S449, the lower allele was deleted in T2 and T3 in patient 2. In patient 4, tumors T11 and T13 demonstrated loss of the lower allele, whereas tumors T7 to T10, and T12 retained heterozygosity with D11S449. Loss of the lower allele in T2 and T3 in patient 2 is seen with marker INT-2. N, matched normal tissue from each patient.
X-Chromosome Inactivation Analysis
Clonality of smooth muscle tumors in two female patients who had three or more different tumors was studied by the X-chromosome inactivation analysis with human androgen receptor (HUMARA), as previously described.21 The technique is based on the random inactivation of one X-chromosome by methylation during female embryogenesis. Tumors derived from a single clone are expected to contain cells with identical allelic methylation. Tumor DNA was extracted and cleaved with the methylation-sensitive restriction endonuclease HpaII (Life Technologies, Gaithersburg, MD). After digestion, the HUMARA locus on the X-chromosome was polymerase chain reaction-amplified using a polymorphic marker. The amplification products were visualized by polyacrylamide gel electrophoresis and autoradiography. The leiomyomatous region was considered to be monoclonal if polymerase chain reaction amplification from HpaII-digested tumor DNA generated a single fragment (upper or lower band) as compared to two fragments of equal intensity in normal polyclonal tissue (Table 2▶ and Figure 2▶ ).
Figure 2.
Representative results of the X-chromosome inactivation analysis of 10 tumors in two female patients. The X-chromosome inactivation method (HUMARA) was used to evaluate clonality of separate tumors. In patient 3, uterine tumor T4 shows methylation of the upper allele and methylation of the lower alleles in uterine tumors T5 and T6. In patient 4, uterine tumor T11 shows methylation of the upper allele, and uterine tumors T12 and T13 show lower allele methylation. At least two of the uterine tumors in each patient arise independently as a different clone. Tumor number corresponds to the tumor number in Table 2▶ . N, normal control, undigested (−) or digested (+), with restriction endonuclease HpaII.
Results
All MEN1-associated and sporadic tumors studied were histologically examined and confirmed as benign leiomyomata.
Fifteen smooth muscle tumors in five female patients with MEN1 included six esophageal, three pulmonary, and six uterine leiomyomata (Table 1)▶ . Four patients had esophageal leiomyomata, two patients had uterine leiomyomata, and one patient had lung leiomyomata. All but one patient had multiple leiomyomata. Patient 4 had tumors in all three sites, whereas all others had tumors limited to a single organ. The age at the diagnosis of MEN1 preceded that of smooth muscle tumor in four patients.
Combined results of LOH on 11q13 and X-chromosome inactivation analysis in the tumors are summarized in Table 2▶ . Ten out of 15 leiomyomata in five patients revealed LOH at the MEN1 gene locus (Table 2▶ , Figure 1▶ ). Multiple tumors from individual patients showed loss of the same allele (upper or lower).
The incidence of 11q13 LOH in MEN1-associated tumors varied by location (Table 2)▶ . Esophageal leiomyomata showed 11q13 LOH in five of six tumors from four patients studied. Similarly, five of six uterine leiomyomata in two patients exhibited 11q13 LOH. All three lung tumors from patient 4 failed to demonstrate 11q13 LOH.
The LOH pattern varied between multiple tumors within an individual patient and suggested that each separate tumor arises from a different clone. In patient 4, the pattern of LOH on 11q13 was different between uterine tumors T12 versus T11 and T13 (Table 2▶ and Figure 1▶ ). Although all three uterine tumors showed LOH with marker PYGM, leiomyomata T11 and T13 showed LOH with D11S449, whereas tumor T12 retained heterozygosity with marker D11S449. Further confirmation of the fact that the multiple uterine leiomyomata in patient 4 developed as separate clones comes from the X-chromosome inactivation analysis. Uterine tumor T11 revealed a single but different clone as compared to tumors T12 and T13 (Table 2▶ and Figure 2▶ ). In patient 3, uterine tumors T4 and T5 had identical 11q13 LOH patterns but demonstrated independent clonal origin by X-chromosome inactivation analysis (Table 2)▶ . Uterine tumor T6 in patient 3 failed to demonstrate 11q13 LOH.
Esophageal leiomyoma T10 in patient 4 showed retention of heterozygosity with both informative markers. An X-chromosome inactivation study showed that the same leiomyoma contained at least two clones (Table 2)▶ . The likely explanation for these results in T10 esophageal tumor is a cross-contamination, ie, two separate clones from the esophageal tumor microdissected and analyzed together, or contamination with normal stromal tissue during microdissection. This effect has been well documented in our previous study3 and has been observed in the previous analysis of the esophageal leiomyoma from patient 1.8 When the same tumor T1 was carefully microdissected and analyzed in the present study, 11q13 LOH with markers D11S480 and INT-2 was documented (Table 2)▶ . In all other cases, X-chromosome inactivation revealed a single clone.
Thirty-nine out of 40 sporadic uterine leiomyomata were informative with at least two markers D11S480, PYGM, or D11S449 at the MEN1 gene locus (Table 3)▶ . One tumor, T32, was not informative with all three markers. 11q13 LOH was evident in only 2 out of 39 (5%) informative sporadic uterine leiomyomata studied. Each of the two tumors exhibited loss with a single microsatellite marker (T23, D11S449, T36, D11S480).
Table 3.
Results of 11q13 LOH in 40 Sporadic Uterine Leiomyomata
| Patient no. | D11S480* | PYGM* | D11S449* |
|---|---|---|---|
| 1 | |||
| 2 | |||
| 3 | |||
| 4 | NI | ||
| 5 | NI | ||
| 6 | X | ||
| 7 | |||
| 8 | |||
| 9 | |||
| 10 | |||
| 11 | |||
| 12 | NI | ||
| 13 | |||
| 14 | |||
| 15 | NI | ||
| 16 | NI | ||
| 17 | NI | ||
| 18 | |||
| 19 | |||
| 20 | |||
| 21 | NI | ||
| 22 | |||
| 23 | • | ||
| 24 | |||
| 25 | NI | ||
| 26 | NI | ||
| 27 | NI | ||
| 28 | |||
| 29 | |||
| 30 | |||
| 31 | |||
| 32 | NI | NI | NI |
| 33 | |||
| 34 | NI | ||
| 35 | |||
| 36 | • | NI | |
| 37 | NI | ||
| 38 | |||
| 39 | |||
| 40 |
•, LOH; , retention of heterozygosity; NI, noninformative; X, did not work.
*Chromosome 11q13 markers are listed in order from centromeric (left) to telomeric (right). MEN1 is between PYGM (CAGA) and D11S449.
Discussion
This study provides insight into the pathogenesis of leiomyomas in MEN1 patients. The two-hit theory of Knudson4 predicts that in a familial tumor syndrome such as MEN1 the genotype of each neoplasm is determined by the presence of the inherited allele with a germline mutation and by the wild-type allele loss through allelic deletion. Molecular genetic studies of MEN1-associated parathyroid, enteropancreatic, and pituitary neoplasms, and mesenchymal tumors, such as lipomas and angiofibromas, have demonstrated LOH at the MEN1 gene region. Here, we report that multiple mesenchymal smooth muscle tumors of the esophagus and uterus in MEN1 patients also develop through the inactivation of the MEN1 gene. Therefore, both esophageal and uterine leiomyomata should be considered an integral part of MEN1. In addition, different LOH patterns on 11q13, in combination with different X-chromosome inactivation patterns, observed in multiple smooth muscle tumors within a single organ and between different organs from the individual MEN1 patient indicate that multiple leiomyomas of the esophagus or uterus arise independently as separate clones. The understanding of these findings should prove useful in the diagnosis and management of MEN1 and in clinical follow-up of MEN1 patients.
The presence of pulmonary leiomyomata has been reported in one MEN1 patient.14 In this study the lung leiomyomata from a single MEN1 patient failed to demonstrate 11q13 LOH with the four markers studied. This may be because of cross-contamination of two tumor clones or undetected small deletions in the MEN1 gene. Alternatively, other mechanisms of allelic inactivation, such as methylation, may play a role in MEN1-associated tumorigenesis. Further studies with larger sample size are necessary to elucidate the role of MEN1 gene in pulmonary smooth muscle tumors.
This is the first report of MEN1 gene involvement in the etiology of uterine leiomyomata in MEN1 patients. All uterine leiomyomata were diagnosed after the diagnosis of MEN1 was established (Table 1)▶ . The late identification of uterine leiomyomata may indicate an altered pathogenesis and growth promotion in MEN1-associated smooth muscle tumors from that found in sporadic tumors, or merely the result of a data collection bias.
Somatic MEN1 gene alterations have been shown to be involved in the pathogenesis of many sporadic counterpart tumors of MEN1.5 The frequency of allelic loss varies with tumor type, which may reflect tissue-specific requirements of the tumor suppressor gene or the presence of more common causative genetic aberrations. Compiled rates of allelic loss are 10 to 18% in sporadic pituitary adenomas, 39% in parathyroid tumors, 51 to 92% in endocrine enteropancreatic tumors, and 60% in carcinoid tumors of the lung and gastrointestinal tract.5,20,22,23 The incidence of 11q13 LOH in sporadic mesenchymal counterpart tumors, however, is uncertain because of small sample size.24,25
Sporadic uterine leiomyomata are the most common neoplasms of the female genital tract affecting 20 to 30% of reproductive age women and accounting for more than one-quarter of all hysterectomies performed in the United States.26,27 Several studies suggested a genetic basis in the tumorigenesis of leiomyomata.28 Approximately 40% of leiomyomata show nonrandom and tumor-specific karyotypic abnormalities that most commonly include: t(12;14)(q15;q23–24), del(7)(q22q32), rearrangements involving 6p21, 10q, trisomy 12, and deletions of 3q.28,29 Structural alterations by cytogenetic analysis have been reported on other chromosomes, including chromosome 11.30 However, no specific gene alterations in the MEN1 gene region have been reported. In our study of 40 sporadic uterine leiomyomata, only two tumors (5%) exhibited 11q13 LOH.
The data suggest that the MEN1 gene contributes to the development of multiple esophageal and uterine leiomyomata in MEN1 patients. In contrast, the gene does not play a significant role in the tumorigenesis of sporadic uterine leiomyomata. Uterine leiomyomata in MEN1 and sporadic patients most likely develop through different pathogenetic mechanisms.
Acknowledgments
We thank Dr. Robert T. Jensen, Digestive Diseases Branch, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, for a referral of one case; and Dr. Denise Peete, National Naval Medical Center, Bethesda, MD, for providing sporadic leiomyoma specimens.
Footnotes
Address reprint requests to Irina A. Lubensky, M.D., Surgical Neurology Branch, National Institute of Neurological Disorders and Stroke, NIH, Building 10, Room 5D37, 10 Center Dr., Bethesda, MD 20892-1414. E-mail: lubenskyi@ninds.nih.gov.
J. L. M. and X. L. equally contributed to this work.
References
- 1.Larsson C, Skogseid B, Öberg K, Nakamura Y, Nordenskjöld M: Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 1988, 332:85-87 [DOI] [PubMed] [Google Scholar]
- 2.Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Speigel AM, Burns AL, Marx SJ: Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276:404-407 [DOI] [PubMed] [Google Scholar]
- 3.Lubensky IA, Debelenko LV, Zhuang Z, Emmert-Buck MR, Dong Q, Chandrasekharappa SC, Guru SC, Manickam P, Olufemi S-E, Marx SJ, Spiegel AM, Collins FS, Liotta LA: Allelic deletions on chromosome 11q13 in multiple tumors from individual MEN1 patients. Cancer Res 1996, 56:5272-5278 [PubMed] [Google Scholar]
- 4.Knudson AG, Jr: Hereditary cancer, oncogenes, and antioncogenes. Cancer Res 1985, 45:1437-1443 [PubMed] [Google Scholar]
- 5.Komminoth P: Review: multiple endocrine neoplasia type 1, sporadic neuroendocrine tumors, and MENIN. Diagn Mol Pathol 1999, 8:107-112 [DOI] [PubMed] [Google Scholar]
- 6.Marx SJ: Multiple endocrine neoplasia type 1. ed 8 Scriver CSet al eds. Metabolic Basis ofInherited Diseases, 2001, :pp 943-966 McGraw Hill, New York [Google Scholar]
- 7.Debelenko LV, Emmert-Buck MR, Zhuang Z, Epshteyn E, Moskaluk CA, Jensen RT, Liotta LA, Lubensky IA: The multiple endocrine neoplasia type I gene locus is involved in the pathogenesis of type II gastric carcinoids. Gastroenterology 1997, 113:773-781 [DOI] [PubMed] [Google Scholar]
- 8.Dong Q, Debelenko LV, Chandrasekharappa SC, Emmert-Buck MR, Zhuang Z, Guru SC, Manickam P, Skarulis M, Lubensky IA, Liotta LA, Collins FS, Marx SJ, Spiegel AM: Loss of heterozygosity at 11q13: analysis of pituitary tumors, lung carcinoids, lipomas, and other uncommon tumors in subjects with familial multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 1997, 82:1416-1420 [DOI] [PubMed] [Google Scholar]
- 9.Pack S, Turner ML, Zhuang Z, Vortmeyer AO, Böni R, Skarulis M, Marx SJ, Darling TN: Cutaneous tumors in patients with multiple endocrine neoplasia type 1 show allelic deletion of the MEN1 gene. J Invest Dermatol 1998, 110:438-441 [DOI] [PubMed] [Google Scholar]
- 10.Vortmeyer AO, Boni R, Pak E, Pack S, Zhuang Z: Multiple endocrine neoplasia 1 gene alterations in MEN1-associated and sporadic lipomas. J Natl Cancer Inst 1998, 90:398-399 [DOI] [PubMed] [Google Scholar]
- 11.Williams ED, Celestin LR: The association of bronchial carcinoid and pluriglandular adenomatosis. Thorax 1962, 17:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berg B, Biorklund A, Grimelius L, Ingemansson S, Larsson L-I, Stenram U, Akerman M: A new pattern of multiple endocrine adenomatosis. Acta Med Scand 1976, 200:321-326 [PubMed] [Google Scholar]
- 13.Burton JL, Hartog M: Multiple endocrine adenomatosis (type 1) with cutaneous leiomyomata and cysts of Moll. Br J Dermatol 1977, 15:S74-S75 [DOI] [PubMed] [Google Scholar]
- 14.Carnevale V, Romagnoli E, D’Erasmo E, Spagna G, Pisani D, Rosso R, Minisola S, Mazzuoli GF: Pulmonary lymphangioleiomyoma in a patient with multiple endocrine neoplasia type 1. J Endocrinol Invest 1997, 20:282-285 [DOI] [PubMed] [Google Scholar]
- 15.Vortmeyer AO, Lubensky IA, Skarulis M, Li G, Moon Y, Park W, Weil R, Barlow C, Spiegel AM, Marx SJ, Zhuang Z: Multiple endocrine neoplasia type 1: atypical presentation, clinical course, and genetic analysis of multiple tumors. Mod Pathol 1999, 12:919-924 [PubMed] [Google Scholar]
- 16.Agarwal SK, Kester MB, Debelenko LV, Heppner C, Emmert-Buck MR, Skarulis MC, Doppman JL, Kim YS, Lubensky IA, Zhuang Z, Green JS, Guru SC, Manickam P, Olufemi S-E, Liotta LA, Chandrasekharappa SC, Collins FS, Spiegel AM, Burns AL, Marx SJ: Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet 1997, 6:1169-1175 [DOI] [PubMed] [Google Scholar]
- 17.Zhuang Z, Bertheau P, Emmert-Buck MR, Liotta LA, Gnarra J, Linehan WM, Lubensky IA: A microdissection technique for archival DNA analysis of specific cell populations in lesions <1 mm in size. Am J Pathol 1995, 146:620-625 [PMC free article] [PubMed] [Google Scholar]
- 18.Manickam P, Guru S, Debelenko LV, Agarwal S, Olufemi S-E, Weisemann J, Boguski MS, Crabtree JS, Wang Y, Roe BA, Lubensky IA, Zhuang Z, Kester MB, Burns AL, Spiegel AM, Marx SJ, Liotta LA, Emmert-Buck MR, Collins FS, Chandrasekharappa SC: Eighteen new polymorphic markers in the multiple endocrine neoplasia type 1 (MEN1) region. Hum Genet 1997, 101:102-108 [DOI] [PubMed] [Google Scholar]
- 19.James MR, Richard CW, III, Schott JJ, Yousry C, Clark K, Bell J, Terwilliger JD, Hazan J, Dubay C, Vignal A, Agrapart M, Imai T, Nakamura Y, Polymeropoulos M, Weissenbach J, Cox DR, Lathrop GM: A radiation hybrid map of 506 STS markers spanning human chromosome 11. Nat Genet 1994, 8:70-76 [DOI] [PubMed] [Google Scholar]
- 20.Debelenko LV, Bambilla E, Agarwal SK, Swalwell JI, Kester MB, Lubensky IA, Zhuang Z, Guru SC, Manickam P, Olufemi SE, Chandrasekharappa SC, Crabtree JS, Kim YS, Heppner C, Burns AL, Spiegel AM, Marx SJ, Liotta LA, Collins FS, Travis WD, Emmert-Buck MR: Identification of MEN1 gene mutation in sporadic carcinoid tumors of the lung. Hum Mol Genet 1997, 6:2285-2290 [DOI] [PubMed] [Google Scholar]
- 21.Zhuang Z, Park W, Pack S, Schmidt L, Vortmeyer AO, Pak E, Pham T, Weil RJ, Candidus S, Lubensky IA, Linehan WM, Zbar B, Weirich G: Trisomy 7-harbouring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nat Genet 1998, 20:66-69 [DOI] [PubMed] [Google Scholar]
- 22.Zhuang Z, Ezzat SZ, Vortmeyer AO, Weil R, Oldfield EH, Park WS, Pack S, Huang S, Agarwal SK, Guru SC, Manickam P, Debelenko LV, Kester MB, Olufemi S-E, Heppner C, Crabtree JS, Burns AL, Spiegel AM, Marx SJ, Chandrasekharappa SC, Collins FS, Emmert-Buck MR, Liotta LA, Asa SL, Lubensky IA: Mutations of the MEN1 tumor suppressor gene in pituitary tumors. Cancer Res 1997, 57:5446-5451 [PubMed] [Google Scholar]
- 23.Boggild MD, Jenkinson S, Pistorello M, Boscaro M, Scanarini M, McTernan P, Perrett CW, Thakker RV, Clayton RN: Molecular genetic studies of sporadic pituitary tumors. J Clin Endocrinol Metab 1994, 78:387-392 [DOI] [PubMed] [Google Scholar]
- 24.Boni R, Vortmeyer AO, Pack S, Park W-S, Burg G, Hofbauer G, Darling T, Liotta L, Zhuang Z: Somatic mutations of the MEN1 tumor suppressor gene detected in sporadic angiofibromas. J Invest Dermatol 1998, 111:539-540 [DOI] [PubMed] [Google Scholar]
- 25.Zhuang Z, Vortmeyer AO, Pack S, Huang S, Pham T, Wang C, Park WS, Agarwal S, Debelenko LV, Kester MB, Guru S, Manickam P, Olufemi S-E, Yu F, Heppner C, Crabtree J, Skarulis M, Venzon DJ, Emmert-Buck MR, Spiegel AM, Chandrasekharappa SC, Collins FS, Burns AL, Marx SJ, Jensen RT, Liotta LA, Lubensky IA: Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res 1997, 57:4682-4686 [PubMed] [Google Scholar]
- 26.Cramer SF, Patel A: The frequency of uterine leiomyomas. Am J Clin Pathol 1990, 94:435-438 [DOI] [PubMed] [Google Scholar]
- 27.Pokras R, Hufnagel VG: Hysterectomies in the United States. Vital Health Stat 1987, 13:1-32 [PubMed] [Google Scholar]
- 28.Ligon AH, Morton CC: Genetics of uterine leiomyomata. Genes Chromosom Cancer 2000, 8:235-245 [PubMed] [Google Scholar]
- 29.Nilbert M, Heim S, Mandahl N, Floderus U-M, Willen H, Mitelman F: Characteristic chromosome abnormalities, including rearrangements of 6p, del(7), +12, and t(12;14), in 44 uterine leiomyomas. Hum Genet 1990, 85:605-611 [DOI] [PubMed] [Google Scholar]
- 30.Mantovani MS, Neto JB, Philbert PM, Casartelli C: Multiple uterine leiomyomas: cytogenetic analysis. Gynecol Oncol 1999, 72:71-75 [DOI] [PubMed] [Google Scholar]