

Abstract
Cells of the soil bacterium Bacillus subtilis have to adapt to fast environmental changes in their natural habitat. Here, we characterized a novel system in which cells respond to heat shock by regulatory proteolysis of a transcriptional repressor CtsR. In B. subtilis, CtsR controls the synthesis of itself, the tyrosine kinase McsB, its activator McsA and the Hsp100/Clp proteins ClpC, ClpE and their cognate peptidase ClpP. The AAA+ protein family members ClpC and ClpE can form an ATP-dependent protease complex with ClpP and are part of the B. subtilis protein quality control system. The regulatory response is mediated by a proteolytic switch, which is formed by these proteins under heat-shock conditions, where the tyrosine kinase McsB acts as a regulated adaptor protein, which in its phosphorylated form activates the Hsp100/Clp protein ClpC and targets the repressor CtsR for degradation by the general protease ClpCP.
Keywords: AAA+ proteins, adaptor proteins, heat shock regulation, proteolysis, tyrosine kinase
Introduction
Cells of the Gram-positive bacterium Bacillus subtilis can respond and adjust to various environmental changes, characteristic of their natural soil habitat. Complex regulatory networks are responsible for the appropriate adaptation to their altered milieu (Price, 2002; Schumann et al, 2002). One of these cellular response systems, the heat-shock regulon, is controlled by at least five different mechanisms, of which the alternative sigma factor σB (Haldenwang and Losick, 1979), the repressors HrcA (Mogk et al, 1997) and CtsR (Krüger and Hecker, 1998; Derre et al, 1999) and the response regulator CssR (Hyyrylainen et al, 2001; Darmon et al, 2002) were identified and characterized as major regulators (Price, 2002; Schumann et al, 2002).
The transcriptional repressor ctsR is the first gene in the clpC operon (Figure 1A). CtsR controls not only transcription of the clpC operon but also of clpP and clpE. The corresponding proteins form part of the B. subtilis protein quality control system, whose function is especially required after heat shock (Krüger et al, 2000; Schlothauer et al, 2003; Gerth et al, 2004).
Figure 1.
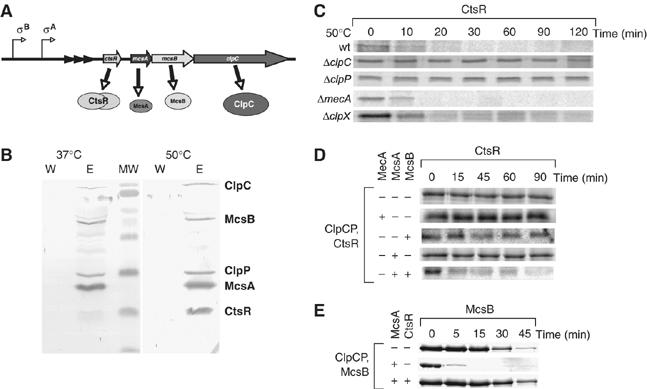
CtsR is targeted for degradation by McsB. (A) Structure of the clpC operon. The triangles in the promoter region represent the CtsR binding sites. (B) An in vivo complex of ClpCP with McsA, McsB and CtsR. An immunoprecipitation with anti-McsB antibodies was performed with B. subtilis 168 cell extracts prepared from exponentially grown cultures at 37°C and one exposed to 50°C for 10 min (as indicated). The last wash step and elution were performed on an SDS–PAGE and the subsequent Western blot developed with anti-ClpC, anti-ClpP, anti-McsB, anti-McsA and anti-CtsR antibody sera is depicted. (C) In vivo stability of CtsR by pulse–chase analysis. Cells of wt, ΔclpC and ΔclpP, ΔmecA, ΔclpC and ΔclpX B. subtilis strains were grown at 37°C and were divided in mid-exponential growth phase and one half was exposed to 50°C. The cells were pulse labeled simultaneously with [35S]methionine for 10 min. Samples were withdrawn at the indicated time points and immunoprecipitated CtsR was detected by autoradiography. (D) In vitro degradation of CtsR. Purified CtsR was incubated in the presence of pyruvate kinase (20 ng/μl), 2 mM PEP and 2 mM ATP with ClpCP and MecA, McsB, McsA, or McsB and McsA (all 1 μM) as indicated in the figure. Samples were taken at the indicated time points, separated on SDS–PAGE and Coomassie-stained CtsR protein is depicted. (E) In vitro degradation of McsB. Purified McsB was incubated in the presence of pyruvate kinase (20 ng/μl), 2 mM PEP and 2 mM ATP with ClpCP and McsA, or McsA and CtsR (all 1 μM) as indicated in the figure. Samples were taken at the indicated time points, separated on SDS–PAGE and the Coomassie-stained McsB protein is depicted.
ClpC and ClpE are members of the Hsp100/Clp proteins of the AAA+ superfamily (Neuwald et al, 1999; Ogura and Wilkinson, 2001; Sauer et al, 2004). Most Hsp100/Clp proteins like ClpA and ClpX from Escherichia coli form hexameric ring-shaped oligomers, which are required for their activity (Horwich et al, 1999; Sauer et al, 2004). They often associate with an oligomeric peptidase like ClpP to form an ATP-dependent protease (Turgay et al, 1998; Gerth et al, 2004), which recognizes, unfolds and translocates substrates, such as misfolded or aggregated proteins, for degradation by ClpP (Horwich et al, 1999; Sauer et al, 2004). ClpA and ClpX can recognize substrate proteins and also employ adaptor proteins like ClpS, SspB or RssB, which enhance and expand substrate repertoire (Becker et al, 1999; Levchenko et al, 2000; Zhou et al, 2001; Dougan et al, 2002; Sauer et al, 2004; Erbse et al, 2006).
ClpC from B. subtilis exhibits unique properties. It not only participates in general proteolysis for protein quality control (Krüger et al, 2000; Schlothauer et al, 2003) but also controls key steps of developmental processes like competence (Turgay et al, 1998), sporulation (Pan et al, 2001) and regulation of stress response (Krüger et al, 2001; Kirstein et al, 2005). It has been proposed that the presence of an adaptor protein, such as MecA, is essential for all the activities of ClpC, as it activates ClpC through a specific interaction with the N-terminal and Linker domains of ClpC, thereby facilitating ClpC oligomerization (Kirstein et al, 2006). A similar activation mechanism was recently proposed for an AAA+ protein involved in multivesicular body sorting in yeast (Azmi et al, 2006).
During the control of regulated proteolysis, the adaptor proteins are able to sense and integrate the signals, which affect the stability of their substrates. One of the first described examples of such a signal-dependent proteolytic switch is the regulation of competence development in B. subtilis. Under normal growth conditions, MecA targets the transcription factor ComK, the master regulator for competence development, for degradation by ClpCP. Competence development is initiated upon a quorum-sensing signal, leading to synthesis of the peptide ComS, whose interaction with MecA results in the release of ComK bringing a halt to the degradation of ComK. In this case, the adaptor protein MecA integrates all aspects of the system, activates ClpCP, targets ComK for degradation and also perceives the signal by interacting with the signaling peptide ComS (D'Souza et al, 1994; Magnuson et al, 1994; Turgay et al, 1997, 1998). Likewise in E. coli, the stability of the alternative sigma factor σS, which regulates the general stress response, is controlled by the adaptor protein RssB (Hengge-Aronis, 2002). RssB, a response regulator, can target σS for degradation by ClpXP (Becker et al, 1999; Zhou et al, 2001). However the stability of σS is controlled by distinct signaling processes, which are regulated by different molecular mechanisms and converge on RssB. Recently, it was demonstrated that phosphorylation of RssB, which is important for the targeting abilities of RssB, is partially controlled by the sensor kinase ArcB, which monitors the cellular energy state (Mika and Hengge, 2005). A more recent study identified a small signaling protein IraP that is synthesized upon phosphate starvation and interacts with RssB, which results in the release of σS from ClpXP-dependent degradation (Bougdour et al, 2006).
In B. subtilis, two other genes of the clpC operon, mcsA and mcsB (Figure 1A), encode for modulators of the activity of CtsR. McsB is a tyrosine kinase, whose activity is induced by McsA, repressed by ClpC and antagonized by the tyrosine phosphatase YwlE. McsB interacts with and phosphorylates tyrosine residues on CtsR, thereby inhibiting its DNA binding activity. We also demonstrated that McsB interacts both with ClpC and CtsR, and was able to compete with MecA for interaction with ClpC, and that both mcsB and mcsA were necessary for the heat-induced degradation of CtsR in vivo (Kirstein et al, 2005).
Based on these results, we hypothesized that McsB could act as an adaptor to activate ClpC and target CtsR for degradation by ClpCP. In this study, we set out to investigate this possibility and provide evidence that McsB, only in its active phosphorylated kinase form, acts as an adaptor protein for ClpC. This new adaptor protein extends the ability of ClpCP to be involved in various regulatory and general proteolysis processes in the B. subtilis physiology.
Results
ClpCP, McsAB and CtsR form a complex under heat-shock conditions in vivo
To examine the ability of McsB to interact with ClpC and CtsR in vivo, co-immunoprecipitation (coIP) experiments were performed using McsB antibodies immobilized to protein A-coated magnetic beads and lysates prepared from wild-type cells either grown at 37°C or heat shocked at 50°C (Figure 1B). Following SDS–PAGE and Western transfer of the separated proteins, immunodetection revealed that ClpC, ClpP, McsA and CtsR were co-immunoprecipitated with McsB under both conditions. Interestingly, the amount of CtsR in the complex was significantly increased after the cells were heat shocked (Figure 1B). This increase in the amount of CtsR, detected under heat-shock conditions, was intriguing, given that there was no dramatic change in the levels of McsA, McsB or ClpCP, and suggested an McsB-mediated interaction of CtsR with ClpCP. We verified these interaction partners of McsB and ClpC independently by mass spectrometry (MS) (data not shown).
This observation raised the question whether McsB, activated by McsA, could directly target CtsR for degradation by ClpCP.
CtsR degradation depends on the presence of ClpCP, McsB and McsA in vivo and in vitro
Previously we demonstrated that under heat-shock conditions, the degradation of CtsR in vivo depended on the presence of McsA and McsB (Kirstein et al, 2005). Using pulse–chase and coIP experiments with anti-CtsR antisera, we have determined the stability of CtsR in clpC, clpP, clpX and mecA deletion strains, demonstrating that neither the ClpC adaptor protein MecA nor the Hsp100/Clp ATPase ClpX (Figure 1C), previously implicated in CtsR degradation (Derre et al, 2000), affect CtsR stability under these conditions. In contrast, ClpC and ClpP are essential for the in vivo degradation of CtsR (Figure 1C).
To further characterize the regulation of CtsR, we reconstituted the ClpCP-dependent degradation of CtsR in vitro (Figure 1D). These experiments demonstrated that CtsR is only efficiently degraded in vitro when both McsA and McsB are present together with the ClpCP protease. ClpCP alone or together with MecA or McsA had no influence on the stability of CtsR (Figure 1D). As McsA was shown to activate kinase activity of McsB and thereby induce phosphorylation of McsB (Kirstein et al, 2005), this result suggests that McsB in its phosphorylated state can act like an adaptor protein, activating ClpC and targeting CtsR for degradation by ClpCP.
It is worth noting that McsB, like MecA, is itself degraded (Figure 1E) and this degradation is enhanced by the addition of McsA, whereas the addition of CtsR was able to stabilize McsB (Figure 1E).
McsB, which exhibits a low autokinase activity, was characterized as an interaction partner for CtsR and ClpC. McsA, on the other hand, was shown to only interact with McsB, thereby activating the kinase activity of McsB (Kirstein et al, 2005). Taken together, our results suggest that only phosphorylated McsB, in its active kinase state, can act as an adaptor protein for ClpC targeting CtsR for degradation by the ClpCP protease.
McsB in the presence of McsA activates the ClpC ATPase
Previously we had shown that the adaptor protein (MecA) was required not only for substrate delivery but also for the formation of ClpC oligomer (Kirstein et al, 2006). Interestingly, the results of the in vitro degradation experiments (Figure 1D and E) suggested that McsB is able to assist the oligomerization of ClpC, which also allowed the assembly of the full proteolytic ClpCP complex and subsequent degradation of CtsR (Figure 1D) or, in the absence of an alternative substrate, McsB (Figure 1E) (Kirstein et al, 2006).
The adaptor protein-induced oligomeric state of ClpC can be easily monitored by measuring the ATPase activity of ClpC (Schlothauer et al, 2003; Kirstein et al, 2006). In the absence of added components, ClpC exhibited little or no ATPase activity, as expected. However, in the presence of McsB and McsA, the ATPase activity of ClpC was fully induced, comparable to the induction by MecA (Figure 2A). Interestingly, addition of McsB alone yielded a comparably weak induction (Figure 2A), whereas McsA alone did not stimulate the ClpC ATPase activity (data not shown). McsB and/or McsA did not display ATPase activity (data not shown). These results suggest that McsB, when activated by McsA, can act as an adaptor protein, which, analogous to the MecA activity, assists the assembly of the ClpC oligomer, thereby enabling the ATPase of ClpC.
Figure 2.
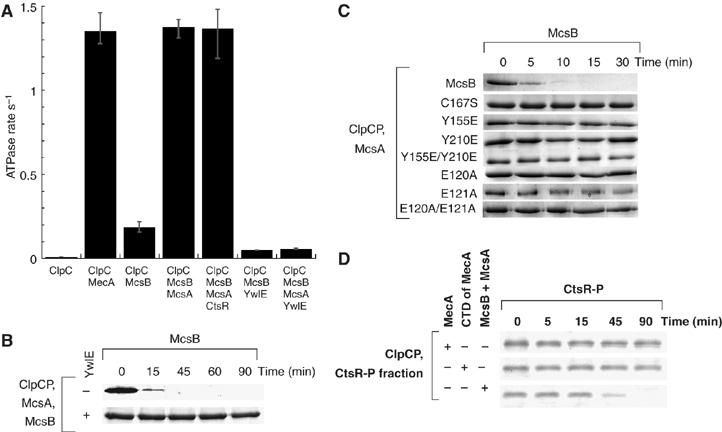
The phosphorylated McsB is the adaptor for ClpC. (A) ATPase activity of ClpC. The ATPase rate of ClpC was determined in the presence of the indicated proteins. (B) In vitro degradation of McsB in the presence of YwlE. The assay was carried out with ClpCP in the presence of McsA, McsB and YwlE as described above. Coomassie-stained McsB is depicted. (C) In vitro degradation of McsB and indicated mutant variants of McsB. The assay was carried out in the presence of McsA, McsB and indicated mutant variants of McsB as described above. Coomassie-stained McsB is depicted. (D) In vitro degradation of CtsR-P. The phosphorylated CtsR fraction obtained as described in Materials and methods was incubated with ClpCP and MecA, the CTD of MecA or McsB and McsA (all 1 μM), as indicated in the figure. Samples were taken at the indicated time points, separated by SDS–PAGE and the Coomassie-stained CtsR protein is depicted.
The adaptor function of McsB is linked to its kinase activity
McsB could act as an adaptor only in the presence of McsA, which is necessary to activate the kinase activity of McsB. We previously identified and characterized YwlE as the cognate phosphatase, which dephosphorylated McsB and McsA (Kirstein et al, 2005). Addition of this phosphatase to the ATPase assay almost completely abolished McsAB-mediated induction of ClpC ATPase (Figure 2A). Consistently, addition of YwlE to ClpCP/McsA in an in vitro degradation assay stabilized McsB (Figure 2B).
To assess whether the kinase activity of McsB is necessary for the adaptor function of McsB, we tested several kinase-deficient variants of McsB. The ability of the mutant McsB proteins to activate ClpC was tested in the presence of McsA by measuring the ATPase activity of ClpC (data not shown) and monitoring ClpCP-dependent degradation of McsB (Figure 2C). Although variants such as McsBY210E, were still able to bind to and inhibit CtsR (Kirstein et al, 2005), none of the McsB variants was able to induce ATPase activity of ClpC (data not shown) or be degraded by ClpCP (Figure 2C) in the presence of McsA.
McsB was fully degraded by ClpCP in the absence of substrate (Figures 1E, 2B and C), suggesting that all McsB proteins were fully phosphorylated in the presence of McsA.
The observed weak activity of McsB in these assays in the absence of McsA (Figures 1C and 2A) can be attributed to the low autokinase activity of McsB (Kirstein et al, 2005).
Taken together, these results demonstrate that the kinase activity of McsB and thereby its phosphorylation status is directly linked to its ability to act as an adaptor protein, assisting the oligomerization of ClpCP and targeting substrate protein to ClpCP.
Phosphorylation of the targeted substrate CtsR is not sufficient for degradation
We previously demonstrated that the McsB kinase phosphorylates CtsR at tyrosine residues (Kirstein et al, 2005). We wanted to examine this process in more detail. Therefore, we generated single point mutant variants of CtsR, where one of the four tyrosine residues of CtsR was replaced by the structural homolog phenylalanine. We analyzed phosphorylation of these variants by the McsB kinase. Our results (Supplementary Figure a) demonstrate that all the variants were still phosphorylated by the kinase, albeit to a decreased level, suggesting that no specific tyrosine is phosphorylated and that CtsR can be phosphorylated at multiple tyrosines in a possibly redundant manner. Although we demonstrated that McsB could activate ClpC and through its interaction with CtsR target CtsR to degradation by ClpCP (Figures 1D and 2A), there is a possibility that the concurrent phosphorylation of CtsR by McsB could mediate the recognition of CtsR-P by ClpC. To test this hypothesis, we phosphorylated CtsR by McsB and McsA in vitro and subsequently separated the phosphorylated CtsR by gel filtration (see Materials and methods and Supplementary Figures b and c). This experiment is also directly demonstrating that McsB, McsA and CtsR are phosphorylated under our experimental conditions (Supplementary Figure c), confirming our previously obtained results (Kirstein et al, 2005).
The fraction of purified and phosphorylated CtsR did not by itself stimulate the ATPase activity of ClpC, excluding the possibility that phosphorylated CtsR could activate ClpC. Then we subjected our obtained fraction of phosphorylated CtsR (Supplementary Figure c) to proteolysis by ClpCP (Figure 2D). The experiment was carried out in the presence of McsB and McsA, MecA or the CTD of MecA, which activates ClpC without targeting substrate, to ensure that CtsR-P can interact with the fully assembled and active ClpCP complex (Kirstein et al, 2006). This experiment, depicted in Figure 2D, demonstrated that the previously phosphorylated CtsR is only degraded in the presence of McsB and McsA. This strongly suggests that the phosphorylation of CtsR is not sufficient to target it for degradation by ClpCP and that CtsR needs to interact with the adaptor McsB-P, which concurrently interacts with ClpC, to be targeted for degradation by ClpCP.
McsB uses the same interaction sites on ClpC as MecA
Interestingly, although McsB and MecA activate ClpC in a similar manner, the two adaptor proteins do not show significant homology (i.e. protein sequence homology is below 20%). Therefore, to further characterize the mechanism of action, we compared the ability of McsB to activate a variety of different ClpC mutants (Figure 3A). For that purpose, we took advantage of experimental approaches, which were applied previously to explore the interaction between ClpC and MecA (Kirstein et al, 2006).
Figure 3.
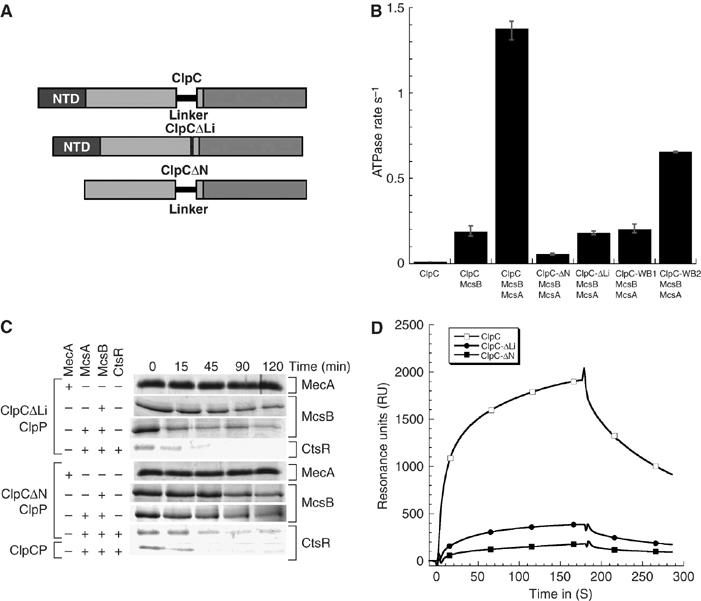
In vitro activity of ClpC lacking its N-terminal domain (ClpC-ΔN) or its Linker domain (ClpC-ΔLi). (A) Domain organization of ClpC, ClpC-ΔLi and ClpC-ΔN. (B) ATPase activity of ClpC and ClpC variants. The ATPase rate of ClpC, ClpC-ΔN, ClpC-ΔLi, ClpC-WB1 or ClpC-WB2 was determined in the presence of the indicated proteins. (C) In vitro degradation assay carried out with ClpC-ΔLi and ClpC-ΔN. The assay was performed as described above. The respective assay components are indicated on the left side and the depicted Coomassie-stained proteins are indicated on the right. (D) Interaction of ClpC, ClpC-ΔLi (both 0.8 μM) and ClpC-ΔN (1.6 μM) with McsB analyzed by SPR. The binding response was measured in resonance units (RU). McsB was immobilized and the indicated proteins were passed over the chip surface.
Initially, we examined ClpC variants in which either the N-terminal domain (ClpC-ΔN) or the Linker domain (ClpC-ΔLi) was deleted (Figure 3A). Interestingly, in contrast to the interaction observed for MecA and ClpC (Kirstein et al, 2006), McsB in the presence of McsA could still induce the ATPase activity of ClpC-ΔLi (Figure 3B).
In addition, McsB (in the presence of ClpC-ΔLi and McsA) was able to target CtsR for degradation by ClpP, with the same efficiency as full-length ClpC (Figure 3C). Conversely, the ATPase activity of ClpC-ΔN was only weakly induced by McsB in the presence of McsA (Figure 3B) and was able to degrade CtsR in the presence of ClpP, albeit at a rate significantly lower than that of ClpC-ΔLi (Figure 3C).
We also measured the interaction of ClpC, ClpC-ΔLi and ClpC-ΔN with immobilized McsB using surface plasmon resonance (SPR) (Figure 3D), reporting the interaction of McsB, which was not activated by McsA, with monomeric ClpC and its variants in the absence of ATP as previously described (Kirstein et al, 2006). These results demonstrate that ClpC-ΔLi shows a significant interaction with McsB, whereas ClpC-ΔN even at the double concentration displayed a weaker but detectable interaction with McsB (Figure 3D).
This suggests that McsB interacts predominantly with the N-terminal domain of ClpC and to a lesser extent with the Linker domain.
We examined the effect of McsB and McsA on the ATPase activity of ClpC protein variants in which either the first or the second Walker B motif (ClpC E280A referred to as ClpC-WB1 and ClpC E618A referred to as ClpC-WB2) were modified (Kirstein et al, 2006). The results depicted in Figure 3B demonstrate that the activity of both single ATPase domains were still induced by McsB in the presence of McsA, albeit less than ClpC. It is interesting to note that the ATPase of ClpC-WB2 was more induced than ClpC-WB1, which is consistent with our finding that the NTD and the Linker domain of ClpC are important interaction sites for McsB, since both domains are located close to the first ATP binding site (Figure 3A), which is still active in ClpC-WB2.
Next, we extended this experimental approach using fusion proteins, where ClpA was used as a scaffold on which its N-terminal domain and/or Linker domain of ClpA were exchanged for those from ClpC (Figure 4A). These hybrid constructs were fully functional as they still exhibited the same basal ATPase rate as ClpA and were able to degrade two characterized ClpA substrates α-casein and ClpA itself (Kirstein et al, 2006). First, we measured the interaction of ClpC, ClpA and the hybrid proteins with immobilized McsB using SPR (Figure 4B) (Kirstein et al, 2006). In these experiments, McsB was able to interact with each ClpA–ClpC hybrid protein to varying extents, but as a control failed to interact with wild-type ClpA. The interaction of McsB with the single-hybrid protein ClpA–CLi was weaker than its interaction with CNTD–ClpA, whereas the interaction with the double-hybrid protein CNTD–ClpA–CLi appeared stronger than the interaction of either single-hybrid proteins (Figure 4B). Interestingly, a strong interaction, with a slow off-rate, was observed for the interaction between McsB and the N-terminal domain of ClpC (Figure 4B), verifying the importance of the N-terminal domain of ClpC for the interaction with McsB. In summary, McsB can interact with ClpC via the N-terminal and the Linker domains of ClpC, but in comparison with MecA (Kirstein et al, 2006), the N-terminal domain of ClpC plays the more dominant role in McsB binding.
Figure 4.
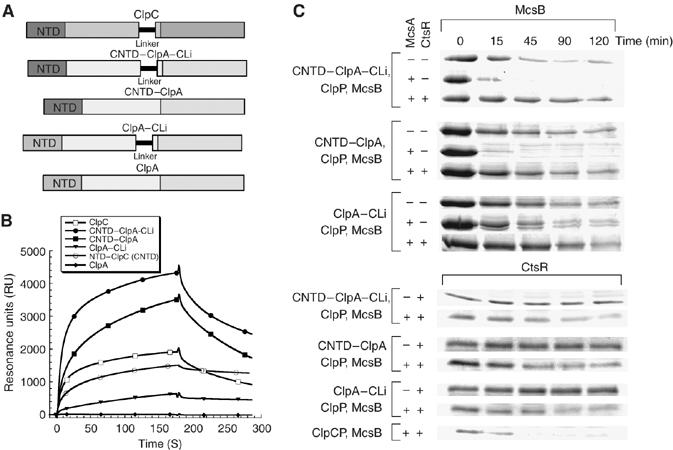
Characterization of the ClpA–ClpC hybrid proteins. (A) Domain organization of ClpC, ClpA and the respective hybrid proteins. (B) Interaction of ClpC, the N-terminal domain of ClpC (CNTD), ClpA and ClpA–ClpC hybrid proteins (CNTD–ClpA, ClpA–CLi and CNTD–ClpA–CLi) (all 0.8 μM) with McsB analyzed by SPR. The binding response was measured in resonance units (RU). McsB was immobilized and the indicated proteins were passed over the chip surface. (C) In vitro degradation assay carried out with the ClpA–ClpC hybrid proteins. The assay was performed as described above. The respective assay components are indicated on the left side and the depicted Coomassie-stained respective proteins McsB and CtsR are indicated above.
To ensure that the interaction, measured using SPR, between these hybrid proteins and McsB was functional, we also analyzed the stability of McsB and CtsR using in vitro degradation assays (Figure 4C). A control experiment demonstrated that McsB was not degraded by ClpAP (data not shown). Importantly, McsB could be degraded by all of the hybrid proteins, although at a slower rate than observed for wild-type ClpC. Nevertheless, the importance of McsA and of the presence of the N-terminal domain of ClpC for a productive interaction with McsB became apparent in these experiments as the degradation was significantly enhanced in the presence of McsA for the hybrid proteins containing the NTD of ClpC. Likewise, CtsR was also degraded by each hybrid protein in a manner similar to that observed for the MecA-dependent degradation of ComK (Kirstein et al, 2006).
In summary, the N-terminal domain seems to play a more important role than the Linker region for the interaction of ClpC with McsB. Although compared with wild-type ClpC, a Linker deletion significantly reduces the induction of the ATPase (Figure 3B) and the Linker domain added to ClpA (ClpA–CLi) still displayed some interaction with McsB and resulted in a low but significant degradation of McsB in the in vitro degradation assay (Figure 4C).
Phosphorylated McsB outcompetes MecA for interaction with ClpC
As both proteins MecA and McsB interacted with the same ClpC domains, we were interested to assess whether ClpC bound one or the other adaptor protein preferentially. For that, the adaptor proteins were incubated at equimolar concentrations with ClpCP and their stability was monitored. In the absence of McsA, both McsB and MecA were degraded with similar kinetics; however, when McsA was added to the reaction, MecA was stabilized significantly (Figure 5A). But this stabilization of MecA was reversed when McsB was replaced with McsBY210E, which is a McsB variant deficient in phosphorylation (Figure 5A). In addition, the unfolded protein α-casein, which is targeted by MecA, could still be degraded by ClpCP/MecA in the presence of McsB, but was stabilized by the additional presence of McsA (Figure 5B). To analyze the relative strength of inhibition by phosphorylated McsB, we monitored the stability of α-casein either in the absence or presence of McsA at a single time point (30 min) and monitored the influence of increasing amounts of MecA (Figure 5C). The amount of α-casein remaining was plotted against MecA concentration (Figure 5C) and from this plot the relative concentration of MecA required to inhibit α-casein degradation was determined. In the presence of McsA, approximately 3 μM MecA was needed to degrade 50% of the α-casein, whereas in its absence, less than 1 μM MecA was sufficient to degrade 50% of the α-casein in 30 min (Figure 5C). These results suggest that under these experimental conditions, the phosphorylated McsB exhibits an apparent higher affinity for ClpC compared with non-phosphorylated McsB and can clearly outcompete MecA even in the presence of its substrate α-casein. The competition is probably mediated mostly by the interaction of McsB with the N-terminal domain of ClpC, which is also an important site for MecA interaction (Kirstein et al, 2006).
Figure 5.
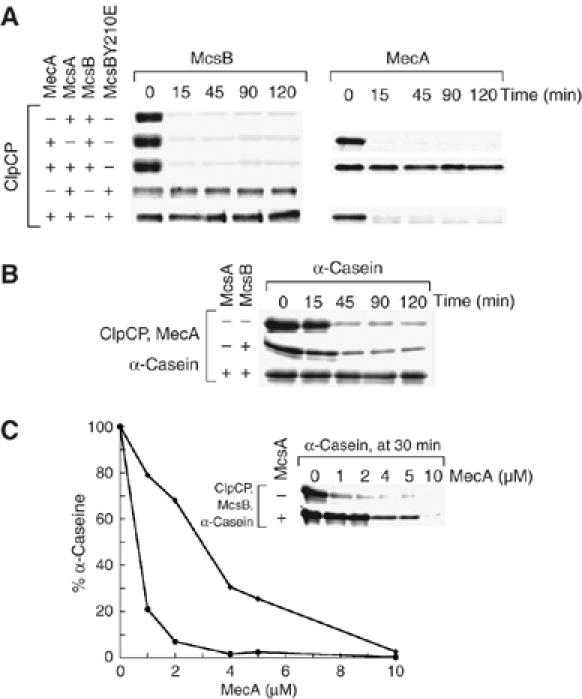
Competition experiments between the adaptor proteins MecA and McsB. (A) In vitro degradation assay including MecA and McsB. The respective assay components are indicated on the left side and the Coomassie-stained proteins MecA and McsB are indicated above. The assay was performed as described above. (B) In vitro degradation assay of α-casein, including MecA and McsB. The respective assay components are indicated on the left side and the Coomassie-stained protein α-casein is indicated above. The assay was performed as described above. (C) In vitro degradation assay of α-casein, including McsB and an increasing amount of MecA. The respective assay components are indicated on the left side and the Coomassie-stained protein α-casein is indicated above. The experiment was carried out as described above, but only the 30 min time point was analyzed and the MecA concentration was varied as indicated below the gel. A graphical representation of the relative amount of α-casein (filled diamonds (⧫) in the presence of McsA and filled circles (•) in the absence of McsA) at different MecA concentrations is also shown.
These competition experiments provide additional evidence that McsB depending on its phosphorylation state acts as an adaptor protein in a comparable manner to MecA, which activates ClpC by assisting its oligomerization and by targeting itself or substrate protein for degradation by ClpCP.
Discussion
In B. subtilis, the controlled degradation of CtsR, which is also the repressor of its own transcription, is an important part of heat-shock regulation (Krüger et al, 2001; Kirstein et al, 2005). Here, we have elucidated the mechanism by which CtsR is specifically targeted for degradation by ClpCP. We have demonstrated that the tyrosine kinase, McsB, when activated by McsA, not only inhibits and phosphorylates CtsR (Kirstein et al, 2005), but also is responsible for degradation and removal of this repressor protein from heat-shocked cells. We provide evidence that McsB, in its activated form, is an adaptor protein for the Hsp100/Clp protein ClpC. Phosphorylated McsB induces ClpC ATPase activity (Figure 2A) and targets the heat-shock repressor CtsR for degradation by ClpCP (Figure 1C and D). In the absence of the substrate (CtsR), McsB is itself degraded by ClpCP, whereas the activator McsA remains stable throughout the course of the experiment (Figure 1E). This behavior is similar to the delivery mechanism described for the adaptor protein MecA (see Table I for a comparison of the adaptor properties of McsB and MecA). Both the functional activation of ClpCP-mediated degradation and the induction of ClpC's ATPase is consistent with a role for McsB in the oligomerization of ClpC (Kirstein et al, 2006).
Table 1.
Comparison of the adaptor properties of MecA and phosphorylated McsB
| Adaptor | Interaction with NTD and Li domain of ClpC | Induction of ClpC ATPase | Degradation in the absence of substrate | Substrates targeted to ClpCP degradation | Regulators of adaptor activity | Expression of adaptor regulated by | Process controlled by proteolysis |
|---|---|---|---|---|---|---|---|
| McsB-P | +a | +a | +a | CtsRa,h,j | McsA, YwlEa | Heat shock, oxidative stress, general stress responsef | Heat shock, oxidative stress and general stress responsef |
| MecA | +b | +b,c | +b,c | ComK, ComSd,h, Spxe,i, α-caseini,j and aggregated proteinc,i | ComSd | Oxidative stressg | Competenced and protein qualityc |
| a This work; b Kirstein et al (2006); c Schlothauer et al (2003); d Turgay et al (1998), e Nakano et al (2002); f Krüger et al (2001); g Leichert et al (2003), Nakano et al (2003); h in vivo and in vitro; i in vitro j phosphorylated. | |||||||
In the absence of McsA, McsB exhibited only limited adaptor function (Figures 1D, 2A, 4C and 5), which suggests that phosphorylation of McsB is important for its adaptor protein function. To confirm the requirement for phosphorylation of McsB, we used variants of McsB, which are unable to be phosphorylated. These mutant proteins were no longer able to carry out the adaptor function of McsB (Figures 2C and 5A). Consistent with these findings, the adaptor activity of McsB was antagonized by the addition of the tyrosine phosphatase YwlE (Figure 2A and B).
The activated kinase McsB is also phosphorylating CtsR; however, concurrent phosphorylation of the substrate CtsR by McsB is not sufficient to target it for degradation by ClpCP (Figure 2D). Therefore, both substrate recognition and concurrent activation of ClpC by McsB, as demonstrated for MecA (Persuh et al, 1999), are necessary for adaptor activity of McsB and targeting of CtsR to ClpCP. Interstingly, α-casein a model substrate for unfolded proteins recognized by MecA is also a phosphorylated protein (Supplementary Figure b), unlike other MecA substrates like ComK, ComS or heat-denatured proteins (Table I), suggesting that phosphorylation of a substrate protein is not directly correlated to its recognition by ClpCP.
In a simple substrate delivery experiment, we were able to show that phosphorylated McsB can inhibit MecA-mediated degradation of α-casein. These data not only confirm that MecA and McsB compete for binding to ClpC but also suggest that phosphorylation of McsB enhances ClpC binding (Figure 5). These results could have important implications in vivo regarding substrate selection by ClpC and are consistent with the observation that the half-life of CtsR is decreased dramatically after heat shock at 50°C (Kirstein et al, 2005). From our data it appears that McsB and MecA use the N-terminal domain of ClpC as a common binding platform. However, in contrast to MecA, the Linker domain of ClpC only plays a supporting role in McsB binding. These results demonstrate that different, not related adaptor proteins can compete with one another for interaction with ClpC, enabling the concurrent involvement of ClpCP in general and regulatory proteolysis, where various and different substrate proteins have to be specifically recognized.
Interestingly, the interaction between McsB and the N-terminal domain of ClpC not only affects McsB-mediated activity of ClpC (Figures 3 and 4) but also affects ClpC-mediated inhibition of McsB kinase activity (data not shown) (Kirstein et al, 2005).
MecA recognizes both general substrates such as unfolded or aggregated proteins (Schlothauer et al, 2003), and specific substrates such as ComK and ComS, by which it is regulated (Turgay et al, 1997, 1998). In comparison with MecA, McsB does not target the unfolded protein α-casein for degradation, suggesting that the general housekeeping functions of the cell are carried out by the MecA–ClpC bi-chaperone system and not by the McsB–ClpC complex (Schlothauer et al, 2003). Rather, McsB only appears to recognize the specific substrate CtsR and is regulated by phosphorylation (see Table I). In general, it appears that specific regulatory substrates like ComK or CtsR are only recognized by their specific and regulated adaptors MecA and McsB to allow precise control of their activity. Nevertheless, the full spectrum of McsB-targeted proteins and the substrate specificity of McsB remain to be seen.
To further understand how ClpC integrates adaptor-mediated general and regulatory proteolyses, detailed structural investigations examining conformational changes of adaptors are required. To that end, it will also be important to elucidate whether the hexameric ClpC ring can interact with multiple adaptors and substrates simultaneously or whether dedicated ClpC–adaptor protein complexes are formed. A recent probabilistic model for the ATPase and substrate binding activity of ClpX (Martin et al, 2005) would suggest that a simultaneous binding of different adaptors with their substrates to ClpC is possible.
Interestingly, a small stabilization of CtsR was also noted in vivo in a clpE deletion and when combined with the clpC mutation, an additional effect on CtsR stability was observed (Miethke et al, 2006), suggesting that ClpE may also be involved in the removal of CtsR. Consistent with our findings, they also demonstrated that deletion of clpC significantly stabilized CtsR levels in vivo. Furthermore, when transcription of all three genes clpE, clpC and clpP is induced, the repressor CtsR must be constantly inactivated and removed to sustain continuous transcription of CtsR controlled genes both under normal and severe stress conditions (Gerth et al, 2004). Taken together, these observations suggest that all proteins encoded by the CtsR regulon are also intimately involved in the proteolytic regulation of their own expression and activity.
Based on our observations that the kinase activity of McsB was inhibited by interaction with ClpC and that McsA activated free McsB, we suggested an enhanced titration model for the regulation of the CtsR activity. We demonstrated that the inhibitory McsB–ClpC interaction could be abolished by a competition with MecA and its substrate α-casein, and that released McsB will be activated and subsequently inhibit CtsR activity (Kirstein et al, 2005). Now that we elucidated the adaptor function of McsB, the model can be expanded to include the regulatory proteolysis of CtsR. Activated McsB will not only inhibit and phosphorylate CtsR, but also target it for degradation by ClpCP. McsB is different from MecA because it appears to interact with ClpC in two different modes depending on its phosphorylation state (Figure 6).
Figure 6.
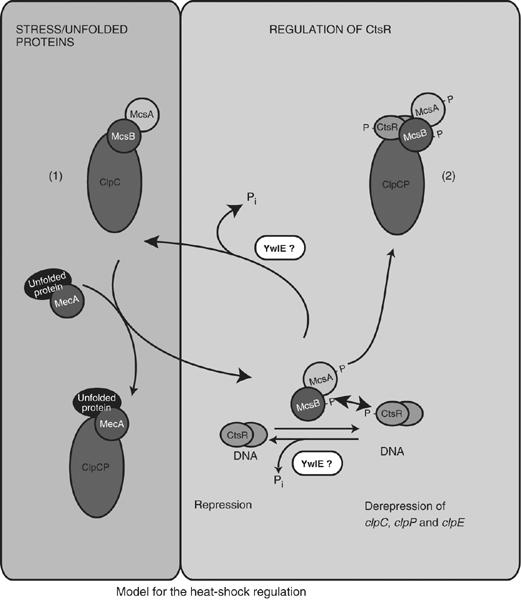
Model of CtsR-mediated heat-shock regulation. The two states of McsB (unphosphorylated, inhibited by ClpC (1) and phosphorylated as active adaptor for ClpCP (2)) are indicated. See Discussion for details.
In the first mode, McsB, in its non-phosphorylated state, interacts with ClpC, which inhibits the kinase activity of McsB. This interaction may already occur when McsB is synthesized cotranslationally with McsA and ClpC. The release of unphosphorylated McsB from ClpC can be triggered by competition with other adaptor proteins, like MecA and YpbH, interacting with unfolded proteins and thereby sensing protein conformational stress ((1) in Figure 6) (Kirstein et al, 2005).
In the second mode, McsB, which is released from the inhibition of its kinase activity, can be activated by McsA, allowing autophosphorylation to occur. The phosphorylated form of McsB acts as an adaptor protein interacting with ClpC in a different mode, as described in this paper ((2) in Figure 6). In this mode, phosphorylated McsB inhibits and phosphorylates CtsR (Kirstein et al, 2005) and ultimately targets CtsR for degradation by ClpCP (Figure 1).
We also determined the in vivo stability of McsB and McsA and observed that both protein species were stable both under normal and heat shock conditions (data not shown). Although a reduced rate of McsB degradation was observed in vitro in the presence of CtsR (Figure 1E) or its cognate phosphatase YwlE (Figure 2B), we suspect that in vivo YwlE might also dephosphorylate free McsB. This activity of YwlE could represent a possible shut-off mechanism to allow the fast return to normal growth conditions after a cessation of the environmental stress (Figure 6).
We demonstrated that CtsR is phosphorylated by the kinase form of McsB (Supplementary figure; Kirstein et al, 2005) and can therefore assume that the degraded CtsR species is also phosphorylated. The two intertangled kinase and adaptor activities are impossible to separate at the moment. Nevertheless, both the modifying by phosphorylation and the proteolytic pathway of CtsR are following the same regulatory logic controlling the CtsR activity (Figure 6). It would be interesting to elucidate in future experiments whether the McsB paralogs in other Gram-positive organisms also exhibit similar adaptor and/or kinase activity.
In summary, we elucidated the mechanism by which the repressor of heat-shock regulation CtsR is removed by regulatory proteolysis from the stressed cells. The identification and characterization of McsB as a new regulated adaptor, which adopts and employs the general protease ClpCP for stress sensing, also illustrates that different adaptor proteins will expand the role of general proteases like ClpCP by conferring distinct substrate specificities for different physiological or regulatory purposes to Hsp100/Clp-mediated proteolysis.
Materials and methods
General methods
DNA manipulations, protein separation and detection were carried out according to standard protocols (Sambrook and Russell, 2001). The construction of the clpC, clpP, clpX (Gerth et al, 1998; Pan et al, 2001) and mecA (Kong and Dubnau, 1994) deletion strain, as well as the antisera were previously described (Gerth et al, 2004; Kirstein et al, 2005). Preparation of soluble cell extracts, co-immunoprecipitation and pulse–chase analysis experiments were performed as described (Kirstein et al, 2005). The co-immunoprecipitated proteins, were in addition to the immunostain (Figure 1B), analyzed and identified by MALDI–TOF-MS at the facilities of the University of Greifswald.
Proteins, cloning, site-directed mutagenesis, expression and purification
Monomer protein concentrations were determined by using the Bio-Rad Bradford assay with BSA as a standard. ClpC, ClpC-ΔLi, ClpC-ΔN, MecA, the CTD of MecA, the ClpC-N-domain, ClpA, CNTD-ClpA, ClpA-CLi, CNTD-ClpA-CLi, ClpC-DWB, ClpC-WB1, ClpCWB2, E. coli and B. subtilis ClpP were expressed and purified as described (Schlothauer et al, 2003; Kirstein et al, 2006). YwlE, McsA, McsB, CtsR, as well as the mutant forms of McsB and CtsR were expressed and purified as described (Kirstein et al, 2005). In addition, McsB was cloned into pQE60 using two primers containing an NcoI and a BamHI cloning site (mcsBpQE60for (5′-CATGCCATGGATGTCGCTAAAGCATTTT ATT CAGG-3′) mcsBpQE60rev (5′-GGGGATCCTATCGATTCATCCTCCTGTCTTTT CCC)). This construct was also used to express and purify McsB. The single Tyr to Phe alanine replacements in CtsR were introduced by site-directed mutagenesis using the oligonucleotides: CtsRY12-Ffor (5′-ATTTCTGACATCATTGAACAATTTTTAAAACG AG-3′), CtsRY12-Frev (5′-TTGTTCAATGATGTCAGAAATATTATGTCC-3′), CtsRY45-Ffor (5′-TGCGTTCCTTCCCAAATAAATTTTGTCATCAA C-3′), CtsRY45-Frev (5′-ATTTATTTGGGAAGGAACGCATTGAAATTT-3′), CtsRY57-Ffor (5′-AGATTTA CAAGCGAAAGAGGATTTATTGTTGA GAGC-3′), CtsRY57-Frev (5′-TCCTCTTTCGCTTGTAAATCTGGTGTTGATG-3′), CtsRY68-Ffor (5′-AGCAAACGCGGGGGCGGCGGTT TTATCAGAATC-3′) and CtsRY68-Frev (5′-ACCGCCGCCCCCGCGTTTGCTCTCAACAAT-3′). Pyruvate kinase and α-casein were purchased from Sigma.
Biochemical assays
The ATPase assays were performed by photometrically measuring the amount of released phosphate, as described in Kirstein et al (2006) and the Biacore analysis was performed on the chip with McsB (3000 RU) immobilized and the ligand concentration was usually 0.8 μM. Only the concentration of ClpC-ΔN was 1.6 μM (Figure 3D), as described in Kirstein et al (2005, 2006). In vitro degradations were performed as described (Kirstein et al, 2006) with protein concentrations of 1 μM, if not stated otherwise. Phosphoenolpyruvate (PEP) and ATP were purchased from Roche. Phosphorylated CtsR was isolated by incubating McsA, McsB and CtsR (each 10 μM) in the presence of ATP (5 mM) at 37°C for 30 min (Kirstein et al, 2005). Subsequently, this mixture was separated by size-exclusion chromatography using a Superose 6 10/300 column (GE Healthcare). The eluted fractions were analyzed by SDS–PAGE and sequentially stained with the Pro-Q Diamond Phosphoprotein Gel Stain system (Molecular Probes) according to the manufacturer's instruction and with colloidal Coomassie. The fraction with pure and phosphorylated CtsR was selected for further ATPase and degradation experiments (Supplementary figure). When, as a control, ClpCP was added to the other fractions with McsB, McsA and CtsR present, CtsR was immediately degraded (data not shown).
Supplementary Material
Supplementary Figures
Acknowledgments
We thank D Dubnau (PHRI, Newark) and A Mogk (ZMBH, Heidelberg) for critical reading of the manuscript. KT thanks R Hengge (FU Berlin) and B Bukau (ZMBH, Heidelberg) for support. KT and MH were supported by the Deutsche Forschungsgemeinschaft.
References
- Azmi I, Davies B, Dimaano C, Payne J, Eckert D, Babst M, Katzmann DJ (2006) Recycling of ESCRTs by the AAA-ATPase Vps4 is regulated by a conserved VSL region in Vta1. J Cell Biol 172: 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker G, Klauck E, Hengge-Aronis R (1999) Regulation of RpoS proteolysis in Escherichia coli: the response regulator RssB is a recognition factor that interacts with the turnover element in RpoS. Proc Natl Acad Sci USA 96: 6439–6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougdour A, Wickner S, Gottesman S (2006) Modulating RssB activity: IraP, a novel regulator of sigma(S) stability in Escherichia coli. Genes Dev 20: 884–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmon E, Noone D, Masson A, Bron S, Kuipers OP, Devine KM, van Dijl JM (2002) A novel class of heat and secretion stress-responsive genes is controlled by the autoregulated CssRS two-component system of Bacillus subtilis. J Bacteriol 184: 5661–5671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derre I, Rapoport G, Msadek T (1999) CtsR, a novel regulator of stress and heat shock response, controls clp and molecular chaperone gene expression in Gram-positive bacteria. Mol Microbiol 31: 117–131 [DOI] [PubMed] [Google Scholar]
- Derre I, Rapoport G, Msadek T (2000) The CtsR regulator of stress response is active as a dimer and specifically degraded in vivo at 37 degrees C. Mol Microbiol 38: 335–347 [DOI] [PubMed] [Google Scholar]
- Dougan D, Mogk A, Zeth K, Turgay K, Bukau B (2002) AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett 529: 6. [DOI] [PubMed] [Google Scholar]
- D'Souza C, Nakano MM, Zuber P (1994) Identification of comS, a gene of the srfA operon that regulates the establishment of genetic competence in Bacillus subtilis. Proc Natl Acad Sci USA 91: 9397–9401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbse A, Schmidt R, Bornemann T, Schneider-Mergener J, Mogk A, Zahn R, Dougan DA, Bukau B (2006) ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature 439: 753–756 [DOI] [PubMed] [Google Scholar]
- Gerth U, Kirstein J, Mostertz J, Waldminghaus T, Miethke M, Kock H, Hecker M (2004) Fine-tuning in regulation of Clp protein content in Bacillus subtilis. J Bacteriol 186: 179–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth U, Krüger E, Derré I, Msadek T, Hecker M (1998) Stress induction of the Bacillus subtilis clpP gene encoding a homologue of the proteolytic component of the Clp protease and the involvement of ClpP and ClpX in stress tolerance. Mol Microbiol 28: 787–802 [DOI] [PubMed] [Google Scholar]
- Haldenwang WG, Losick R (1979) A modified RNA polymerase transcribes a cloned gene under sporulation control in Bacillus subtilis. Nature 282: 256–260 [DOI] [PubMed] [Google Scholar]
- Hengge-Aronis R (2002) Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev 66: 373–395, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwich AL, Weber-Ban EU, Finley D (1999) Chaperone rings in protein folding and degradation. Proc Natl Acad Sci USA 96: 11033–11040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyyrylainen HL, Bolhuis A, Darmon E, Muukkonen L, Koski P, Vitikainen M, Sarvas M, Pragai Z, Bron S, van Dijl JM, Kontinen VP (2001) A novel two-component regulatory system in Bacillus subtilis for the survival of severe secretion stress. Mol Microbiol 41: 1159–1172 [DOI] [PubMed] [Google Scholar]
- Kirstein J, Schlothauer T, Dougan DA, Lilie H, Tischendorf G, Mogk A, Bukau B, Turgay K (2006) Adaptor protein controlled oligomerization activates the AAA+ protein ClpC. EMBO J 25: 1481–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein J, Zuhlke D, Gerth U, Turgay K, Hecker M (2005) A tyrosine kinase and its activator control the activity of the CtsR heat shock repressor in B. subtilis. EMBO J 24: 3435–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Dubnau D (1994) Regulation of competence-specific gene expression by Mec-mediated protein–protein interaction in Bacillus subtilis. Proc Natl Acad Sci USA 91: 5793–5797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E, Hecker M (1998) The first gene of the Bacillus subtilis clpC operon, ctsR, encodes a negative regulator of its own operon and other class III heat shock genes. J Bacteriol 180: 6681–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E, Witt E, Ohlmeier S, Hanschke R, Hecker M (2000) The clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J Bacteriol 182: 3259–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger E, Zühlke D, Witt E, Ludwig H, Hecker M (2001) Clp-mediated proteolysis in Gram-positive bacteria is autoregulated by the stability of a repressor. EMBO J 20: 852–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leichert LI, Scharf C, Hecker M (2003) Global characterization of disulfide stress in Bacillus subtilis. J Bacteriol 185: 1967–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Seidel M, Sauer RT, Baker TA (2000) A specificity-enhancing factor for the ClpXP degradation machine. Science 289: 2354–2356 [DOI] [PubMed] [Google Scholar]
- Magnuson R, Solomon J, Grossman AD (1994) Biochemical and genetic characterization of a competence pheromone from B. subtilis. Cell 77: 207–216 [DOI] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT (2005) Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature 437: 1115–1120 [DOI] [PubMed] [Google Scholar]
- Miethke M, Hecker M, Gerth U (2006) Involvement of Bacillus subtilis ClpE in CtsR degradation and protein quality control. J Bacteriol 188: 4610–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika F, Hengge R (2005) A two-component phosphotransfer network involving ArcB, ArcA, and RssB coordinates synthesis and proteolysis of sigmaS (RpoS) in E. coli. Genes Dev 19: 2770–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Homuth G, Scholz C, Kim L, Schmid FX, Schumann W (1997) The GroE chaperonin machine is a major modulator of the CIRCE heat shock regulon of Bacillus subtilis. EMBO J 16: 4579–4590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S, Kuster-Schock E, Grossman AD, Zuber P (2003) Spx-dependent global transcriptional control is induced by thiol-specific oxidative stress in Bacillus subtilis. Proc Natl Acad Sci USA 100: 13603–13608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S, Zheng G, Nakano MM, Zuber P (2002) Multiple pathways of Spx (YjbD) proteolysis in Bacillus subtilis. J Bacteriol 184: 3664–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV (1999) AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9: 27–43 [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure-diverse function. Genes Cells 6: 575–597 [DOI] [PubMed] [Google Scholar]
- Pan Q, Garsin DA, Losick R (2001) Self-reinforcing activation of a cell-specific transcription factor by proteolysis of an anti-sigma Factor in B. subtilis. Mol Cell 8: 873–883 [DOI] [PubMed] [Google Scholar]
- Persuh M, Turgay K, Mandic-Mulec I, Dubnau D (1999) The N- and C-terminal domains of MecA recognize different partners in the competence molecular switch. Mol Microbiol 33: 886–894 [DOI] [PubMed] [Google Scholar]
- Price CW (2002) General stress response. In Bacillus subtilis and its Closest Relatives : From Genes to Cells, Sonenshein AL, Hoch JA, Losick R (eds) pp 369–384. Washington, DC: ASM Press [Google Scholar]
- Sambrook J, Russell D (2001) Molecular Cloning A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ES, Siddiqui SM, Wah DA, Baker TA (2004) Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119: 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlothauer T, Mogk A, Dougan DA, Bukau B, Turgay K (2003) MecA, an adaptor protein necessary for ClpC chaperone activity. Proc Natl Acad Sci USA 100: 2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann W, Hecker M, Msadek T (2002) Regulation and function of heat-inducible genes in Bacillus subtilis. In Bacillus subtilis and its Closest Relatives: From Genes to Cells, Sonenshein AL, Hoch JA, Losick R (eds) Washington, DC: ASM Press [Google Scholar]
- Turgay K, Hahn J, Burghoorn J, Dubnau D (1998) Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J 17: 6730–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgay K, Hamoen LW, Venema G, Dubnau D (1997) Biochemical characterization of a molecular switch involving the heat shock protein ClpC, which controls the activity of ComK, the competence transcription factor of Bacillus subtilis. Genes Dev 11: 119–128 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Gottesman S, Hoskins JR, Maurizi MR, Wickner S (2001) The RssB response regulator directly targets sigma(S) for degradation by ClpXP. Genes Dev 15: 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures