

Abstract
Aspartylglycosaminuria (AGU) is one of the most common lysosomal storage disorders in humans. A mouse model for AGU has been recently generated through targeted disruption of the glycosylasparaginase gene, and at a young age the glycosyl asparaginase-deficient mice demonstrated many pathological changes found in human AGU patients (Kaartinen V, Mononen I, Voncken J-W, Gonzalez-Gomez I, Heisterkamp N, Groffen J: A mouse model for aspartylglycosaminuria. Nat Med 1996, 2:1375–1378). Our current findings demonstrate that after the age of 10 months, the general condition of null mutant mice gradually deteriorated. They suffered from a progressive motoric impairment and impaired bladder function and died prematurely. A widespread lysosomal hypertrophy in the central nervous system was detected. This neuronal vacuolation was particularly severe in the lateral thalamic nuclei, medullary reticular nuclei, vestibular nuclei, inferior olivary complex, and deep cerebellar nuclei. The oldest animals (20 months old) displayed a clear neuronal loss and gliosis, particularly in those regions, where the most severe vacuolation was found. The severe ataxic gait of the older mice was likely due to the dramatic loss of Purkinje cells, intensive astrogliosis and vacuolation of neurons in the deep cerebellar nuclei, and the severe vacuolation of the cells in vestibular and cochlear nuclei. The impaired bladder function and subsequent hydronephrosis were secondary to involvement of the central nervous system. These findings demonstrate that the glycosylasparaginase-deficient mice share many neuropathological features with human AGU patients, providing a suitable animal model to test therapeutic strategies in the treatment of the central nervous system effects in AGU.
Aspartylglycosaminuria (AGU), which is the most common disorder of glycoprotein degradation, is an autosomal recessively inherited lysosomal storage disease. 1,2 AGU is caused by mutations in the glycosylasparaginase gene, which result in deficient glycosylasparaginase activity. Recent isolation and characterization of human glycosylasparaginase and cloning and sequencing of its cDNA revealed that glycosylasparaginase is a 346-amino acid residue glycoprotein. 3 The precursor polypeptide undergoes a unique posttranslational autocatalytic activation step in the endoplasmic reticulum. 4 The generated subunits are further processed in lysosomes, in which a mature, biologically active heterotetrameric glycosylasparaginase is formed. 5
Glycosylasparaginase is a ubiquitously expressed enzyme, which hydrolyzes the N-glycosidic linkage between N-acetylglucosamine and l-asparagine during the lysosomal degradation of Asn-linked glycoproteins. 6 In AGU, as a consequence of lacking glycosylasparaginase activity, aspartylglucosamine and other glycoasparagines accumulate in tissues and body fluids. 7 Histological findings include hypertrophied storage lysosomes, which can be found in multiple tissues and cell types. 8,9
The clinical course of AGU is characterized by a relatively normal early development followed by the progressive psychomotor degeneration. 10 Other clinical manifestations include coarse facial features, short stature, and connective tissue lesions. The exact pathophysiology of AGU, is not known and currently there is no therapy for this ultimately fatal disease. Recently, we generated a mouse model for AGU using gene targeting and embryo transfer techniques. 11 Here, we report novel neuropathological findings of the glycosylasparaginase-deficient mice, which demonstrate that in many aspects, this mouse model accurately mimics the human disease. Moreover, the results presented in this article explain some unique phenotypic features of the glycosylasparaginase-deficient mice.
Materials and Methods
Animals
The glycosylasparaginase gene was disrupted in R1 embryonic stem cells by homologous recombination, and the cells were injected into recipient blastocysts from C57BL/6J mice as described. 11 Two independently targeted clones were used to generate the null mice. Mice were genotyped by Southern blot analysis of tail DNA collected from pups at 10 days of age as described. 11 Footprint analyses were carried out as described. 12 All mice were housed under standard conditions.
Pathology
For gross pathology, mice were euthanized and necropsies were performed. For histopathological analysis, tissues were fixed by intracardiac perfusion with 10% buffered formalin or 2.5% glutaraldehyde. The brain, spinal cord, and visceral organs were removed, processed, and embedded in paraffin or epoxy resin. Paraffin-embedded tissues were cut to 5 μm and stained with hematoxylin and eosin, whereas epoxy resin-embedded tissues were cut to 1 μm and stained with toluidine blue. A total of three knockout and three wild-type control mice were used for each histological study. Urine specimens were analyzed for bacterial growth using standard procedures at the Department of Pathology and Laboratory Medicine, Childrens Hospital Los Angeles.
Immunohistochemistry
The sections were deparaffinized and hydrated, and the endogenous peroxidase activity was eliminated by incubating the sections with 3% H2O2 for 30 minutes. Immunostaining was done using a HistoMouse kit (Zymed Laboratories Inc., San Francisco, CA) according to the manufacturer’s instructions. Monoclonal rat anti-glial fibrillary acidic protein (GFAP) antibody also was from Zymed.
Results
Lifespan and External Phenotypes
Early development of the AGU mice was indistinguishable from that of their wild-type littermates. 11 First external signs of deterioration were noticed at approximately 5 months of age, when the null mutant mice started to display a general scruffiness, easily recognizable from the disheveled coat. First spontaneous deaths among null mutants occurred at the age of 10 months, and the average lifespan of AGU mice was 19 months (n = 6), which is significantly shorter than the average lifespan of their wild-type littermates (>28 months; n = 10). All of the AGU mice that reached the age of 18 months or more had significant difficulty moving, and they had poor balance and coordination. The steps were short, and the gait was often straddle legged (Figure 1 ▶ , lane 3). In addition, AGU mice were often dragging their hind legs while they were walking, which can also be seen in a footprint assay (Figure 1 ▶ , lane 3).
Figure 1.

AGU mice show an abnormal gait pattern. Hind footprint patterns of two AGU mice at 22 months of age (lanes 2 and 3) were compared with the footprint pattern of a wild-type littermate (lane 1). The AGU mouse (lane 2) walked with steps two times shorter than those of the control mouse (lane 1). In addition to short steps, the knockout mouse (lane 3) was walking straddle-legged, dragging its hind limbs.
Histology of the Central Nervous System
Because AGU is mainly a neurological disease, we studied the histopathology of the central nervous system (CNS) in detail. Whereas gross external examination showed no differences in the CNS between the controls and knockouts, more detailed studies revealed several impairments typical for AGU mice.
Cytoplasmic Vacuolation
In the null mutant mice, progressive cytoplasmic vacuolation could be clearly detected (Tables 1 and 2) ▶ ▶ . In the cerebral cortex, fine or coarse cytoplasmic vacuoles were found in a moderate number of neurons, particularly in those of the lower cortical layers (Table 1 ▶ , Figure 2 ▶ ). In general, the small neurons were much less affected than large neurons. In the hippocampus of the 6-month-old glycosylasparaginase (−/−) mice, granular cells of the dentate gyrus appeared to be unaffected, whereas the larger neurons of the Amnon’s horn displayed some hypertrophic storage lysosomes (Table 1) ▶ . In the few surviving older glycosylasparaginase-deficient mice (>20 months old), all of the neurons both in the dentate gyrus and Amnon’s horn displayed moderate to severe vacuolation. The thalamus showed marked neuronal vacuolation, particularly in the lateral nuclei (Table 1) ▶ . In contrast, the neuronal population of the basal ganglia appeared to be only slightly vacuolated. In the brainstem, all of the major neuronal aggregates displayed moderate to severe cytoplasmic vacuolation (Table 2) ▶ . Similar to the supratentorial structures, the large neurons were more severely affected. Particularly involved were the tegmental nuclei and substantia nigra, medullary reticular nuclei, vestibular and cochlear nuclei, and inferior olivary complex (Table 2) ▶ . The neurons of the deep cerebellar nuclei showed massive vacuolation (Figure 2B) ▶ , whereas in the cerebellar cortex, Purkinje cells were less affected, and the internal granular cells did not show any detectable vacuolation (Table 2) ▶ . In addition to neurons, glial cells in the glycosylasparaginase-deficient mice contained hypertrophic storage lysosomes. However, this vacuolation was fine and less extensive than in neurons.
Table 1.
Distribution of Vacuolized Cells in the CNS of Glycosylasparaginase-Deficient Mice (n = 3 in Each Age Group) at 6 Months, 10 Months, and 20 Months of Age (Cerebral Cortex, Hippocampus, Thalamus, Hypothalamus, Amygdala Complex, Septal Nuclei, and Caudate/Putamen)
| Location | Amount | ||
|---|---|---|---|
| 6 months | 10 months | 20 months | |
| Cerebral cortex | |||
| Cingulate | ++ | ++ | +++ |
| Frontal, parietal | ++ | ++ | +++ |
| Insular, orbital | ++ | ++ | +++ |
| Piriform | + | ++ | +++ |
| Claustrum | + | + | +++ |
| Entorhinal | + | ++ | +++ |
| Subiculum | + | ++ | +++ |
| Occipital | ++ | ++ | +++ |
| Hippocampus | |||
| Amnon’s horn | + | ++ | +++ |
| Dentate gyrus | − | ++ | ++ |
| Hilus | ++ | +++ | ++++ |
| Thalamus | |||
| Anterior thalamic nuclei | + | ++ | ++ |
| Medial thalamic nuclei | ++ | ++ | +++ |
| Lateral thalamic nuclei | +++ | +++ | ++++ |
| Subthalamic nuclei | +++ | +++ | +++ |
| Medial habenular nuclei | − | ++ | +++ |
| Lateral habenular nuclei | ++ | ++ | ++ |
| Hypothalamus | ++ | +++ | +++ |
| Amygdala complex | ++ | +++ | +++ |
| Septal nuclei | + | ++ | +++ |
| Caudate/putamen | + | ++ | +++ |
++++, very large amount of heavily vacuolized neurons; +++, large amount of vacuolized neurons; ++, moderate amount of vacuolized neurons; +, small amount of vacuolized neurons; −, no vacuolized neurons.
Table 2.
Distribution of Vacuolized Cells in the CNS of Glycosylasparaginase-Deficient Mice (n = 3) at 6 Months, 10 Months, and 20 Months of Age (Cerebellum, Midbrain, Pons, and Medulla)
| Location | Amount | ||
|---|---|---|---|
| 6 months | 10 months | 20 months | |
| Cerebellum | |||
| Interposed cerebellar nuclei | +++ | +++ | ++++ |
| Lateral cerebellar nuclei | +++ | +++ | ++++ |
| Medial cerebellar nuclei | +++ | +++ | ++++ |
| Vermis | |||
| Purkinje cells | + | ↓+ | ↓+ |
| Granular cells | − | − | − |
| Hemisphere | |||
| Purkinje cells | + | ↓+ | ↓++ |
| Granular cells | − | − | − |
| Flocculus | |||
| Purkinje cells | + | ↓+ | ↓+ |
| Granular cells | − | − | − |
| Midbrain | |||
| Inferior colliculus | ++ | ++ | +++ |
| Superior colliculus | ++ | +++ | +++ |
| Periaqueductal gray | + | +++ | +++ |
| IV nucleus | ++ | ++ | +++ |
| Paralemniscal nuclei | ++ | ++ | ++ |
| Pararubrospinal nuclei | ++ | ++ | ++ |
| Interpeduncluar nuclei | ++ | ++ | +++ |
| Red nucleus | +++ | +++ | +++ |
| Substantia nigra | ++ | ++ | +++ |
| Pons | |||
| Facilial nucleus | ++ | ++ | +++ |
| Abducens nucleus | − | + | ++ |
| Pontine reticular nucleus | ++ | ++ | ++++ |
| Superior olive | + | ++ | ++ |
| Trapezoid body | + | ++ | ++ |
| Trigeminal nuclei | ++ | ++ | +++ |
| Locus ceroleus | ++ | ++ | ++ |
| Basis pontis nuclei | + | +++ | +++ |
| Supragenual nucleus | ++ | ++ | ++ |
| Medulla | |||
| Spinal trigminal nucleus | ++ | +++ | ++++ |
| Medullary reticular nucleus (ventral) | +++ | +++ | ++++ |
| Medullary reticular nucleus (dorsal) | +++ | +++ | ++++ |
| Reticular complex | ++ | ++ | +++ |
| Cuneatus nucleus | +++ | +++ | +++ |
| Gracilis nucleus | ++ | +++ | +++ |
| Solitary nucleus | +++ | +++ | +++ |
| Dorsal cochlear nucleus | +++ | +++ | +++ |
| Vestibular nuclei | +++ | +++ | +++ |
| Tractus solitary nuclei | ++ | ++ | ++++ |
| Inferior olivary complex | +++ | +++ | ++++ |
++++, very large amount of heavily vacuolized neurons; +++, large amount of vacuolized neurons; ++, moderate amount of vacuolized neurons; +, small amount of vacuolized neurons; −, no vacuolized neurons; ↓, extensive cell loss.
Figure 2.

Neurons in the CNS of the glycosylasparaginase-deficient mice demonstrate extensive cytoplasmic vacuolization. A: A transverse section through the isocortex shows extensive cytoplasmic vacuolization (arrows), which involves the neurons of all layers (toluidine blue staining; semithin 1-μm section; magnification, ×250). B: A section of the deep cerebellar nucleus displays clusters of large neurons separated by tortuous myelinated fibers. The fine vacuoles are dispersed throughout the cytoplasm of the neurons (arrows) (toluidine blue staining; semithin 1-μm section; magnification, ×250).
Neuronal Loss and Astrogliosis
In addition to severe neuronal vacuolation, all older AGU mice (>18 months) displayed a scattered neuronal loss, which was severe in the lower cortical neurons and in the medial and lateral thalamic nuclei. In the cerebellar cortex, the loss of Purkinje cells was striking (70 to 80%) when compared with age-matched controls (Figure 3, A and B) ▶ . Immunostaining with GFAP antibody demonstrated significant gliosis with hypertrophic astrocytes in areas where profound neuronal loss and vacuolation could be detected, such as in the thalamic lateral nuclei (Figure 4, C and D) ▶ . Moreover, hypertrophic astrocytes could be seen in the subcortical white matter, white matter tracts, and commissures (Figure 4, A and B) ▶ in the null mutant mice. In the cerebellum, remarkable gliosis and hypertrophic astrocytes were demonstrated in the deep cerebellar white matter and between neurons in the deep cerebellar nuclei (Figure 3, E and F) ▶ . The molecular layer of the knockout cerebellum also displayed a clear increase of the GFAP-positive Bergmann glial radial processes (Figure 3, C and D) ▶ . In concordance with the neuronal loss, the bulk of white matter was smaller in the AGU mice than in controls (data not shown).
Figure 3.
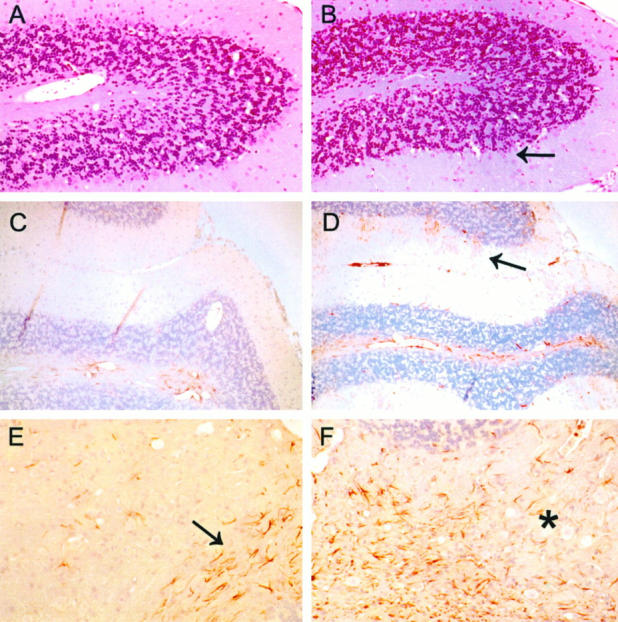
Cerebellum is severely affected in the AGU mice. A knockout cerebellar lobe (B) shows a complete loss of Purkinje cells (arrow), whereas an age-matched control displays a normal number of Purkinje cells (A). Knockout sections also demonstrate a clear increase in the number of GFAP-positive Bergmann glial processes (red) in the molecular layer (D, arrow) when compared with a wild-type control (C). In a wild-type section (E), the normal focal meshwork of GFAP-positive glia is limited to the subcortical white matter near the fourth ventricle (arrow), whereas a knockout displays a marked gliosis (asterisk) with hypertrophic astrocytes extending into the cerebellar white matter and between neurons of the cerebellar nuclei (F). A and B, H&E staining; magnification, ×100. C through F, immunostaining for GFAP; magnification, ×100.
Figure 4.

Extensive gliosis in the CNS of the glycosylasparaginase-deficient mice. The glial density and size of their cytoplasmic processes (red staining) is markedly increased throughout the corpus callosum and deep white matter in AGU mice 22 months old (B). For comparison, the wild-type control (A) shows a moderate number of astrocytes evenly distributed along the corpus callosum (asterisk) and few isolated astrocytes or glial fibrils in the deep white matter (star). In the wild-type sample, only a few isolated astrocytes can be seen in thalamus (lateral nuclei) (C), whereas a knockout sample displays a marked proliferation of hypertrophic astrocytes with long processes (D). Immunostaining for GFAP; magnification, ×100.
Terminally Ill Mice
All AGU mice that reached the age of 18 months or more suffered from severe ataxia, as described above. Another consistent phenotype of these mice was a failure to urinate, which led to a massive expansion of the bladder (approximately 30 times the volume of a normal filled bladder), as shown in Figure 5 ▶ . This phenotype was already detectable at the age of 5 to 6 months, and it showed slow but steady progression, culminating in extreme bladder dilation, as shown in Figure 5 ▶ . As a consequence, the urothelium was stretched to a thickness of two cells in the most distended area (Figure 6, A and B) ▶ . However, the peripheral nerves around the neck of the expanded bladder showed no pathological changes (Figure 6, C and D) ▶ , suggesting that this phenotype was caused by the degeneration of the CNS. As a consequence of the marked bladder dilation, kidneys showed clear signs of hydronephrosis as demonstrated by tubular atrophy, glomerular loss, and interstitial fibrosis (Figure 7A) ▶ . The urine inside dilatated bladders was clear, and analyses for bacterial growth were negative, clearly demonstrating that this phenotype was not due to bacterial infections. Many terminally ill AGU mice also suffered from massive coagulative hepatic necrosis (Figure 7B) ▶ , with the residual hepatocytes forming an irregular periportal rim. Moreover, a band of acute inflammatory infiltrate could be seen at the interface between residual and necrotic parenchyma, as well as sinusoidal fibrin thrombi and occasional organizing thrombi in portal and central veins.
Figure 5.
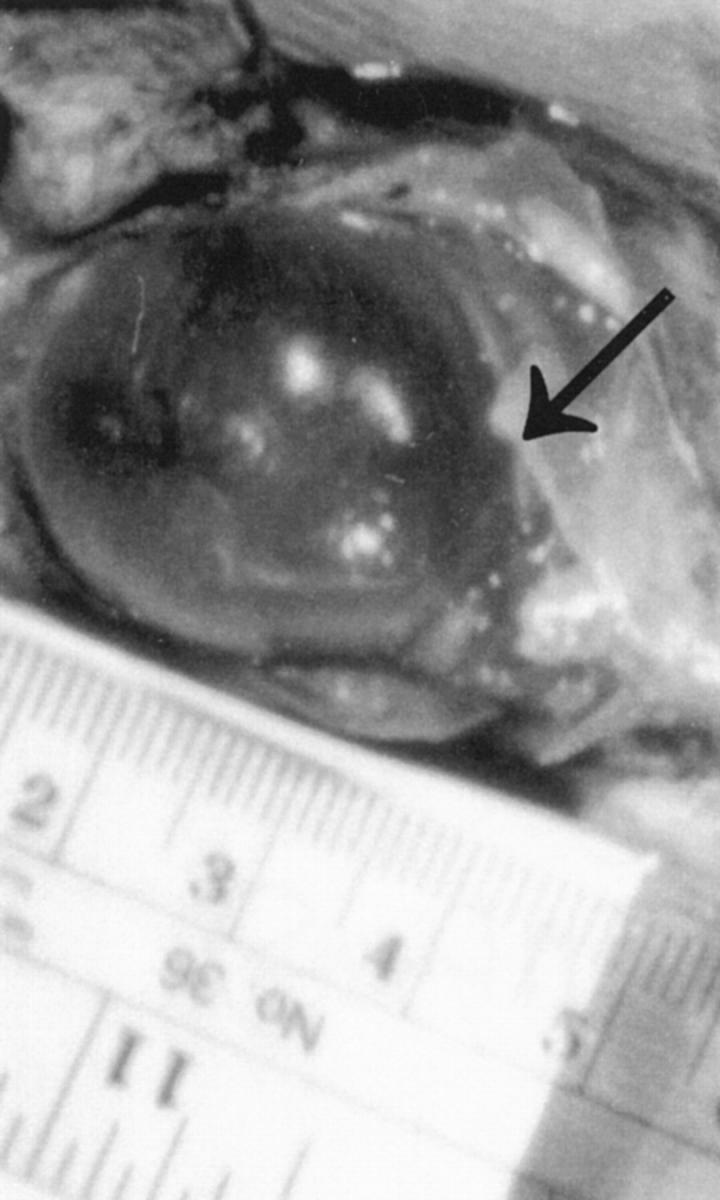
AGU mice display massively swollen urinary bladders that lack detectable neuronal abnormalities. The diameter of the bladder of this female mouse (26 g) at the age of 18 months exceeded 3 cm.
Figure 6.

Urinary bladder of the glycosylasparaginase-deficient mouse lacks detectable neuronal abnormalities. Full-thickness section of the urinary bladder of a distended (A, fundus) and less distended (B, neck) area of the bladder wall. The thickness of the urothelium varies with the degree of bladder distention, stretching to a thickness of two cells in the most distended area (arrow). The lamina propria and muscularis are also thin, with the latter reduced to a few thin parallel fascicles. C: Section through the serosa of the urinary bladder. A portion of a normal autonomic ganglion (asterisk) and adjacent nerve fibers (arrow) are shown. The endothelial cells of a vascular sinusoid (arrowhead) and mesothelial cells (star) are also normal. D: Transverse section through the bladder neck showing normal unmyelinated nerve twigs (arrow) and blood vessels in a loose connective tissue stroma. Scattered fibroblasts exhibit foamy cytoplasmic changes (asterisk). Magnification: A and B, ×100; C and D, ×250.
Figure 7.
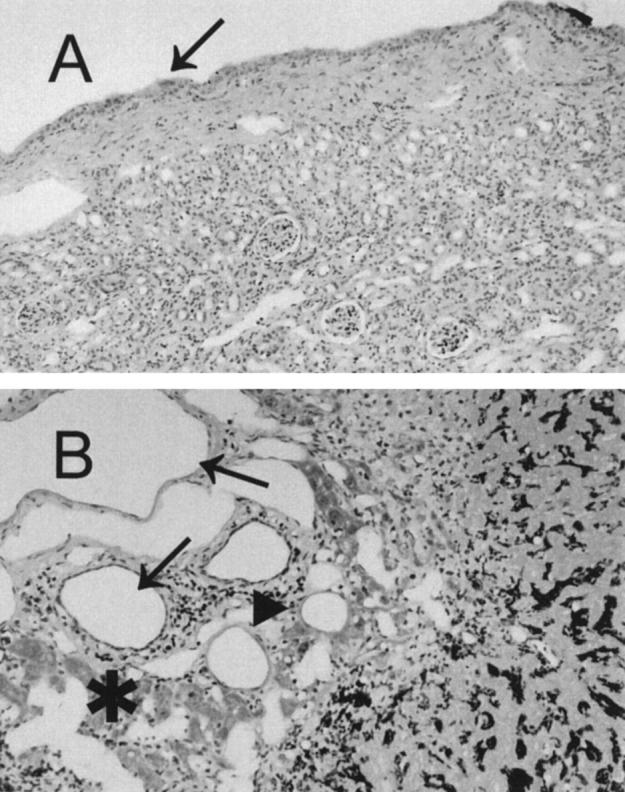
Terminally ill AGU mice display pathological changes in kidney (A) and liver (B). A: Renal parenchyma is thin with associated glomerular loss, tubular atrophy, and interstitial fibrosis. The caliceal mucosa is distended to one or two urothelial cells (arrow). B: The portal tract is expanded because of marked dilation of the terminal sinusoids and portal vein (arrows). Most of the hepatic parenchyma has undergone coagulative necrosis; there is only an irregular periportal rim of hepatocytes remaining (asterisk). A band of acute inflammatory infiltrate is present at the interface between residual and necrotic parenchyma (arrowhead). Magnification, ×40.
Discussion
Previously, we have demonstrated that mice homozygous for the disrupted glycosylasparaginase locus exhibit a complete absence of glycosylasparaginase activity, which leads to the accumulation of aspartylglucosamine in their tissues and urine. 11 In the present study, we analyzed the neuropathology of the AGU mice in detail. The results presented here will be important in assessment of the value of the glycosylasparaginase-deficient mouse as a model for the human AGU disease. Moreover, the detailed histological characterization of these knockout mice is an important step in understanding the etiology of this progressive neurodegenerative disease.
Histological studies of tissues of human AGU patients have shown that hypertrophic storage lysosomes can be found both in the CNS as well as in visceral organs. 8,9 It has been reported that in the CNS, the cerebral cortex, thalamus, and substantia nigra are all heavily affected. In the cerebellum, a diffuse loss of Purkinje cells has been reported as well as some proliferation and hypertrophy of Bergmann glia. 9 The dentate, red, inferior olivary, pontine, and cranial nerve nuclei of the brainstem have been found to be less affected. Unlike the CNS, visceral organs such as kidneys and liver of human AGU patients are able to maintain their functionality despite the extensive vacuolation of cells.
As our present results show, glycosylasparaginase-deficient mice share many similarities with human AGU patients. Like the human AGU patients, the null mutant mice also display widely distributed vacuolation of cells both in the CNS and in visceral organs, astrogliosis in the CNS, shortened lifespan, and motoric impairment. Some features of AGU mice are unique and have not been reported in humans. Our results clearly demonstrate that in AGU mice, neuronal vacuolation is a progressive event and that in old mice, severe vacuolation is often associated with a neuronal loss. All of the aging AGU mice (>18 months) suffered from impaired bladder function, often associated with a reflux nephropathy. Because the peripheral nerves were unaffected, we concluded that this “neurogenic bladder” phenotype was presumably secondary to the CNS involvement.
Older mice (>18 months) showed a severe ataxic gait. This was likely due to the significant damage found in the cerebellum, as demonstrated by the dramatic loss of Purkinje cells, intensive astrogliosis, and vacuolation of neurons in deep cerebellar nuclei. Especially the loss of Purkinje cells, which form excitatory synapses with parallel and climbing fibers and play a prominent role in motor plasticity, may have largely contributed to this phenotype. Unlike in human AGU patients, for whom scattered loss of Purkinje cells has been described, in AGU mice the loss of Purkinje cells is widespread. In addition to the damage in the cerebellum, the severe vacuolation of cells in vestibular and cochlear nuclei may have contributed to the development of this motoric phenotype as well.
Human AGU patients die prematurely. The expected life span is most often less than 45 years, bacterial infections being the most common cause of death. 10 The hepatic necrosis found in some of the terminally ill AGU mice was also caused by bacterial infections, and therefore it is likely that the loss of glycosylasparaginase activity both in humans and in mice impairs defense mechanisms toward bacterial infections.
Our present results further confirm our previous data that the glycosylasparaginase-deficient mouse develops a progressive disease with biochemical and histopathological hallmarks of human AGU, and therefore it can be used as a model for this disease. An interesting question remains whether the vacuolation of cells in the CNS is enough to cause the development of this progressive neurodegenerative disease or whether the loss of glycosylasparaginase will also influence other, currently unknown aspects of metabolism in the CNS. The finding that both the neuronal loss and astrogliosis colocalize with the neuronal vacuolation suggests that accumulation of hypertrophic storage vacuoles is capable of inducing pronounced neuronal damage, and thus neuronal vacuolation alone could significantly contribute to the development of AGU. The glycosylasparaginase-deficient mice provide a valuable tool for further characterization of the pathogenesis of AGU and testing the efficacy of different therapeutic approaches such as gene therapy and enzyme replacement therapy to reverse the CNS changes in AGU.
Footnotes
Address reprint requests to Vesa Kaartinen, Department of Pathology, MS no. 103, Childrens Hospital Los Angeles, 4650 Sunset Boulevard, Los Angeles, CA 90027.
Supported by Sigrid Juselius Foundation and Pediatric Research Foundation (Ulla Hjelt Fund) (to IM).
References
- 1.Thomas GH, Beaudet AL: Disorders of glycoprotein degradation and structure: α-mannosidosis, sialidosis, aspartylglucosaminuria, and carbohydrate-deficient glycoprotein syndrome. The Metabolic and Molecular Bases of Inherited Disease. Edited by CR Scriver, AL Beaudet, WS Sly, D Valle. New York, McGraw-Hill, 1995, pp 2545–2548
- 2.Mononen I, Fisher KJ, Kaartinen V, Aronson NN, Jr: Aspartylglycosaminuria: protein chemistry and molecular biology of the most common lysosomal storage disorder of glycoprotein degradation. FASEB J 1993, 7:1247-1256 [DOI] [PubMed] [Google Scholar]
- 3.Aronson NN Jr: Molecular biology of glycosylasparaginase. Lysosomal Storage Disease: Aspartylglycosaminuria. Edited by I Mononen, NN Aronson Jr. Austin, TX, R. G. Landes Co./Heidelberg, Springer-Verlag, 1997, pp 119–136
- 4.Ikonen E, Julkunen I, Tollersrud O-K, Kalkkinen N, Peltonen L: Lysosomal aspartylglucosaminidase is processed to the active subunit complex in the endoplasmic reticulum. EMBO J 1993, 12:295-302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guan C, Cui T, Rao V, Liao W, Benner J, Lin CL, Comb D: Activation of glycosylasparaginase: formation of active N-terminal threonine by intramolecular autoproteolysis. J Biol Chem 1996, 271:1732-1737 [DOI] [PubMed] [Google Scholar]
- 6.Aronson NN Jr: Asparagine-linked glycoproteins and their degradation. Lysosomal Storage Disease: Aspartylglycosaminuria. Edited by I Mononen, NN Aronson Jr. Austin, TX, R. G. Landes Co./Heidelberg, Springer-Verlag, 1997, pp 55–75
- 7.Mononen T, Mononen I: Biochemistry and biochemical diagrams of aspartylglycosaminuria. Edited by I Mononen, NN Aronson Jr. Austin, TX, R. G. Landes Co./Heidelberg, Springer-Verlag, 1997, pp 41–49
- 8.Arstila AU, Palo J, Haltia, Riekkinen P, Autio S: Aspartylglucosaminuria. I. Fine structural studies of liver, kidney and brain. Acta Neuropathol 1972, 20:207–216 [DOI] [PubMed]
- 9.Haltia M, Palo J, Autio S: Aspartylglucosaminuria: a generalized storage disease. Acta Neuropathol 1975, 31:243-255 [DOI] [PubMed] [Google Scholar]
- 10.Arvio M, Autio S, Mononen T: Clinical manifestations of aspartylglycosaminuria. Lysosomal Storage Disease: Aspartylglycosaminuria. Edited by I Mononen, NN Aronson Jr. Austin, TX, R. G. Landes Co./Heidelberg, Springer-Verlag, 1997, pp 55–75.
- 11.Kaartinen V, Mononen I, Voncken J-W, Gonzalez-Gomez I, Heisterkamp N, Groffen J: A mouse model for aspartylglycosaminuria. Nat Med 1996, 2:1375-1378 [DOI] [PubMed] [Google Scholar]
- 12.Aida A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S: Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 1994, 79:377-388 [PubMed] [Google Scholar]