

Abstract
The pH- and electrolyte-dependent charging of collagen I fibrils was analyzed by streaming potential/streaming current experiments using the Microslit Electrokinetic Setup. Differential scanning calorimetry and circular dichroism spectroscopy were applied in similar electrolyte solutions to characterize the influence of electrostatic interactions on the conformational stability of the protein. The acid base behavior of collagen I was found to be strongly influenced by the ionic strength in KCl as well as in CaCl2 solutions. An increase of the ionic strength with KCl from 10−4 M to 10−2 M shifts the isoelectric point (IEP) of the protein from pH 7.5 to 5.3. However, a similar increase of the ionic strength in CaCl2 solutions shifts the IEP from 7.5 to above pH 9. Enhanced thermal stability with increasing ionic strength was observed by differential scanning calorimetry in both electrolyte systems. In line with this, circular dichroism spectroscopy results show an increase of the helicity with increasing ionic strength. Better screening of charged residues and the formation of salt bridges are assumed to cause the stabilization of collagen I with increasing ionic strength in both electrolyte systems. Preferential adsorption of hydroxide ions onto intrinsically uncharged sites in KCl solutions and calcium binding to negatively charged carboxylic acid moieties in CaCl2 solutions are concluded to shift the IEP and influence the conformational stability of the protein.
INTRODUCTION
Collagen type I is the most abundant fibrillar protein in the extracellular matrix of connective tissues and receives highest attention in matrix biology. Technologies as different as the design of morphogenetic matrices for regenerative therapies (1) and leather processing (2–4) essentially depend on a detailed understanding of structure-property relations of this key molecule and its assemblies. Despite the huge progress achieved in the recent past, more information about environmental factors influencing structural transitions of collagen I is required—for a rigorous explanation of the protein's functionality as well as for improved processing of collagen-based materials.
The basic structure of the collagen molecule is the right-handed triple helix (5–8). The ability of collagen I to form fibrils is highly dependent on the structure and conformation of this triple helix (9–12). Three left-handed supercoiled polypeptide chains (two a1(I) and one a2(I)) of a polyproline-II-like conformation form this right-handed triple helix with a diameter of 1.5 nm and a length of 300 nm (5–8). Every chain contains the repeating unit Gly-X-Y and the small glycine on every third position allows the close packing of the triple helix. The X and the Y positions are highly exposed to the solvent (13,14), where the glycine residues are facing each other in the interior of the triple helix (5,8). In addition to the high amount of Pro and Hyp (which is well known to stabilize the triple helix conformation) a high quantity of ionizable residues (14) (∼15–20% of all residues (15,16)) is present in every polypeptide chain. Therefore, electrostatic interactions should obviously be relevant for the stability of the triple helix conformation as well as for biological specificity and fibril formation (13,16–18).
In the past, the influence of the electrolyte composition of aqueous media on the stability of collagen was mostly addressed by thermoanalytical studies. It was shown that thermal denaturation of collagen leads to the destruction of the triple helix into a randomly coiled form in which the three chains are separated (19). It was shown recently that the denaturation temperature of collagen depends on the scan rate (20,21), therefore scanning rates should be considered when results of different authors are discussed. Furthermore the thermal stability of soluble collagen molecules or collagen aggregates was found to depend strongly on the solution pH (22–26), i.e., on the status of dissociation of the ionizable amino-acid residues. By changing the solution pH at invariant electrolyte background a maximum thermal stability of the collagen I molecules was determined in the physiological pH range (22).
The importance of pH and ionic strength for the thermal stability of collagen has been convincingly demonstrated by several authors. For varied electrolytes, the thermal stability of collagen I correlates with both the type of salt and the solution pH (22,26). For CaCl2 a decrease in thermal stability of soluble collagen I with increasing salt concentration was observed in the physiological pH range while the thermal stability was reported to increase with increasing salt concentration from 0.4 to 1 M at pH = 2.3 (22). In the case of KCl at pH ∼6 a slight decrease of the thermal stability was found with increasing salt at low concentrations (from 0 to 0.2 M), yet a further increase of the salt concentration was shown to increase the thermal stability as well (22). However, at pH = 2.3 the stability is decreased with increasing electrolyte concentration up to a KCl concentration of 0.5 M (22). Brown et al. (27) and Komsa-Penkova et al. (28) report a slightly decreased stability of collagen with increased KCl concentration up to 0.4 M at pH 3; further increase of the concentration was found to result in a stabilization. Arktas (26) reported an increase of the thermal stability of insoluble connective tissues with the increase of the ionic strength of NaCl from 0 to ∼1.02 M at pH 3.7 and pH 5.7, but a decrease of the thermal stability was observed for the addition of a similar amount of CaCl2 at pH 5.7.
Obviously, the pH of the solution is a very important parameter for the thermal stability and conformation of collagen molecules in different electrolytes. However, previous reports either concern soluble collagen I in the acidic pH range (27,28) or insoluble aggregates (tendons, tissues) at a wider pH range (23–26). In addition, all authors (except for (28)) applied pure, deionized water and solutions of ∼10−1 M (or even higher) as the lowest salt concentration. Beyond that, the salt concentration of the solution provides a means to control the screening of charges: The range of electrostatic interactions is characterized by the Debye screening length κ−1 (see Eq. 1), the characteristic distance over which the electrostatic potential near a charged entity decays,
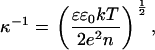 |
(1) |
where ɛ is the relative permittivity (dielectric constant) of the solution, ɛ0 the permittivity, k the Boltzmann constant, T the absolute temperature, e the elementary charge, and n = NA × I ×1000 is the number concentration (with NA Avogadro constant and I the ionic strength of the solution). Therefore κ−1 is reduced from a theoretical maximum of ∼950 nm in pure water to a range of 0.3 nm at the ionic strength of I = 1 M at 298.15 K. Thus, electrostatic interactions stabilizing or destabilizing the conformation of collagen can be expected to be rather strong at low ionic strengths. Although living tissues are characterized by rather high ionic strengths (e.g., PBS, 0.154 M NaCl buffered with Na2HPO4/KH2PO4, ionic strength = 0.16 M) and low Debye screening lengths (0.75 nm at the conditions given), overemphasizing and modulating electrostatics in solutions of lower and varied ionic strength can help to accentuate the interactions responsible for the structure of the protein and the influence of the binding of certain ions can be studied in a more straightforward fashion.
The work reported below therefore investigated the electrostatic interactions influencing the conformation and stability of collagen I at screening lengths varied over several orders of magnitude, at varied pH values and in the presence of various selected electrolytes. A simple 1:1 electrolyte (KCl) was applied to modulate the screening of charged groups only, furthermore divalent positive ions (Ca2+) were used to investigate possible preferential interactions with charged amino-acid side chains at the collagen molecule. To address the above-mentioned issues we analyzed collagen I samples with differential scanning calorimetry (DSC) and circular dichroism spectroscopy (CD). These studies were complemented by monitoring the ionization of the protein in situ as a function of the composition of aqueous environments using a novel electrokinetic approach (29), which was applied to collagen-coated substrates developed for this particular experiment. The results of this electrokinetic analysis were found to correlate well with the measurements of conformation and thermal stability (for a set of similar solutions), revealing the important role of electrostatic interactions in the modulation of structural features of collagen I.
MATERIALS AND METHODS
Preparation of collagen samples for DSC and CD measurements
Sample type A
The collagen solution (sterile, pepsin-solubilized bovine dermal collagen I solution in 0.012 N HCl, Vitrogen, Cohesion Technologies, Palo Alto, CA, 3.0 mg/mL) was diluted with the different salt solutions to 0.6 mg/mL and dialyzed in Visking dialysis tubes (MWCO 14000, Carl Roth, Karlsruhe, Germany) five times (each time for 12 h) against the corresponding electrolyte solutions. The dialysis was monitored by measuring the conductivity in the supernatant after 12 h. During the last two dialyses steps, the conductivity of the supernatant was equal to the theoretical conductivity of the designated electrolyte solutions. The resulting solutions were homogenized for 60 s (T-8 Ultra Turrax, IKA, Staufen, Germany) and subsequently analyzed by DSC and CD-spectroscopy. The pH of the solutions was checked and found to be ∼pH = 6 throughout the experiments.
Sample type B
Alternatively, fibril formation of a collagen sample was induced before the dialysis. Eight parts of the acidic collagen solution (Vitrogen, 3.0 mg/mL) were mixed with one part of 10-fold concentrated PBS (Sigma, Steinheim, Germany) and one part of 0.1 M NaOH (Merck, Darmstadt, Germany) (temperature was kept at ∼2–5°C by using an ice bath). The pH was checked to be pH 7.4 (cColl = 2.4 mg/mL). Before filling the dialysis tubes, the solutions were diluted with PBS to 0.6 mg/mL. After incubation of the dialysis tubes (in 37°C PBS solution) in a CO2-free incubator for 12 h, the tubes were removed from the PBS solution and dialyzed against the different electrolyte solutions as described above. The resulting solutions were homogenized for 60 s (T-8 Ultra Turrax) before any other measurement; the solution pH was found to be close to pH = 6 for all samples.
Preparation of collagen-coated substrates for electrokinetic measurements
Freshly cleaned planar glass (polished glass carriers 10 mm × 20 mm, Herbert Kubatz, Berlin, Germany) or silicon oxide carrier materials (silicon wafers 10 mm × 20 mm, GeSiM mbH Grosserkmannsdorf, Germany) (oxidized in a mixture of aqueous ammonia solution (Acros Organics, Geel, Belgium) and hydrogen peroxide (Merck)) were functionalized by reaction with 3-aminopropyl-dimethylethoxy-silane (ABCR, Karlsruhe, Germany). Poly(octadecene-alt-maleic anhydride) MW 30,000–50,000 (Polysciences, Warrington, PA) was dissolved in tetrahydrofuran (THF, Fluka, Deisenhofen, Germany) in a concentration of 0.15 wt % and used for spin coating (RC 5 Suess Microtec, Garching, Germany) of the amine-modified carriers at 4000 rpm for 30 s. The obtained copolymer coatings (freshly tempered at 120°C for 2 h) were used for the deposition of reconstituted fibrillar collagen from collagen I solution already mixed with PBS (Sigma) and 0.1 M NaOH (Merck) corresponding to the protocol for the sample preparation (see sample type B) for DSC and CD measurements. The carriers were immersed in cell culture dishes (Nunc, Wiesbaden, Germany) in 4 mL of the further diluted (chilled PBS, Sigma) collagen solution at a collagen concentration of 0.6 mg/mL. Fibrils were formed by increasing the temperature to 37°C in an incubator. After keeping at 37°C for 12 h in the incubator the carriers were removed and washed excessively in PBS and three times in MilliQ water (deionized, decarbonized water, R = 18.2 MΩ cm; Millipore, Billerica, MA). The resulting thin collagen layers on top of the glass carriers were air-dried and stored in a closed box. The collagen-covered surfaces were characterized by atomic force microscopy and by scanning electron microscopy. Atomic force microscopy measurements (Bioscope, Veeco, Santa Barbara, CA) were accomplished on collagen samples in potassium chloride solutions in tapping mode with a Point Probe silica cantilever (Nanosensors, Wetzlar, Germany; ∼10 nm tip radius). For the scanning electron microscope (XL 30 ESEM FEG, FEI-Philips, Eindhoven, Netherlands) air-dried samples were gold-coated with a sputter coater (SCD 050, BAL-TEC, Schalksmühle, Germany) and investigated in the high vacuum mode at an acceleration voltage of 5–10 kV. For further information on the preparation and characteristics of the collagen/maleic anhydride copolymer films, the reader is referred to the literature (30,31).
Differential scanning calorimetry (DSC)
DSC thermograms of 0.6 mg/mL collagen preparations in different electrolyte solutions were recorded in a micro-DSC III heat conduction scanning microcalorimeter (Setaram, Caluire, France) with a sample volume of 750 mL. The same volume of the appropriate electrolyte solution was taken as a blank sample. The 1-mL ampoules (Hastelloy C, High Performance Alloys, Windfall, IN) were allowed to stabilize at 25°C before initiation of the scanning experiment over a temperature range of 25–110°C. The heating rate was 0.5 K/min for all experiments.
The enthalpy of transition (ΔdH) was calculated from the area of the peak. The apparent melting temperature (denaturation temperature, Td) was defined as the temperature at which a local maximum occurs in the heat excess capacity. Although a heating rate of 0.5 K/min does not produce an equilibrium-melting curve (20,23), the transition approximates a two-state model and the apparent melting temperature values obtained under these conditions are useful for comparison. Average values of at least three measurements are reported.
Circular dichroism (CD) spectroscopy
CD spectra were measured at room temperature (23°C ± 1°C, thermostated) with a J 810 spectropolarimeter (JASCO, Gross-Umstadt, Germany) equipped with a 150 W Xenon lamp and cuvettes with 1 mm path length. CD spectra were recorded from 280 to 180 nm with a scanning speed of 50 nm/min. All spectra were background-corrected (by measuring the correlating electrolyte solutions without collagen) and average values of at least four independent measurements were considered. To avoid protein damage, cumulative exposure of any sample did not exceed 8 min per sample. The ready dialyzed collagen solutions were measured at concentrations of 0.6 mg/mL and further diluted before the measurement at 0.3 mg/mL and 0.15 mg/mL, respectively. CD spectra were collected in mdeq and converted to molar ellipticity (see Table 1) by using the equation
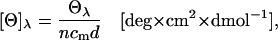 |
(2) |
where n is the number of amino-acid residues in the protein chain (3045 residues according to HPLC data for Vitrogen (27)), cm the molar concentration in mol/L (M), and d the pathlength in mm. The molecular weight used for calculation was 285,000 g/mol (27).
TABLE 1.
CD data of collagen samples dialyzed against different electrolyte solutions
| Sample
|
Min
|
Max
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| I* [M] | ccoll† [mg/mL] | λ [nm] | CD [mdeq] ± 6.5% | CD [deg cm2 dmol−1] ± 6.5% | Cross λ [nm] | λ [nm] | CD [mdeq]± 6.5% | CD [deg cm2 dmol−1] ± 6.5% | Rpn‡ ± 0.01 |
| 10−4 KCl | 0.15 | 197 | −90.2 | −56289 | 213 | 221 | 11.5 | 7157 | 0.13 |
| 10−4 KCl | 0.30 | 197 | −186.6 | −58229 | 213 | 221 | 23.3 | 7279 | 0.13 |
| 10−4 KCl | 0.60 | — | — | — | 213 | 221 | 45.9 | 7152 | 0.13§ |
| 10−3 KCl | 0.15 | 198 | −66.2 | −41228 | 213 | 221 | 10.8 | 6764 | 0.16 |
| 10−3 KCl | 0.30 | 198 | −145.3 | −45326 | 213 | 221 | 22.4 | 6982 | 0.15 |
| 10−3 KCl | 0.60 | — | — | — | 213 | 221 | 44.9 | 7009 | 0.16§ |
| 10−2 KCl | 0.15 | 202 | −17.0 | −10601 | 215 | 223 | 7.2 | 4474 | 0.42 |
| 10−2 KCl | 0.30 | 202 | −35.4 | −11029 | 215 | 223 | 15.3 | 4783 | 0.43 |
| 10−2 KCl | 0.60 | 202 | −66.8 | −10416 | 215 | 223 | 28.6 | 4468 | 0.43 |
| 10−4 CaCl2 | 0.15 | 197 | −93.2 | −58129 | 213 | 221 | 12.8 | 8006 | 0.14 |
| 10−4 CaCl2 | 0.30 | 197 | −180.4 | −56273 | 213 | 221 | 26.0 | 8102 | 0.14 |
| 10−4 CaCl2 | 0.60 | — | — | — | 213 | 221 | 53.0 | 8268 | 0.14§ |
| 10−3 CaCl2 | 0.15 | 198 | −72.4 | −45157 | 213 | 221 | 11.4 | 7101 | 0.16 |
| 10−3 CaCl2 | 0.30 | 198 | −139.5 | −43507 | 213 | 221 | 22.8 | 7126 | 0.16 |
| 10−3 CaCl2 | 0.60 | — | — | — | 213 | 221 | 45.9 | 7166 | 0.16§ |
| 10−2 CaCl2 | 0.15 | 200 | −34.6 | −21577 | 214 | 222 | 9.2 | 5728 | 0.27 |
| 10−2 CaCl2 | 0.30 | 200 | −63.7 | −19858 | 214 | 222 | 17.4 | 5429 | 0.27 |
| 10−2 CaCl2 | 0.60 | 200 | −122.1 | −19048 | 214 | 222 | 33.2 | 5176 | 0.27 |
Ionic strength of the sample.
Collagen concentration of the samples. Only the 0.6 mg/mL sample was dialyzed over five days; the other two concentrations were diluted with the electrolyte solution before the CD measurement. Measurements were performed at 23°C.
Rpn represents the ratio of positive peak intensity over negative peak intensity (absolute values).
Rpn values at given ionic strengths are assumed to be constant because of the linear increase of the CD signal in the 221 nm region (positive peak) for all collagen concentrations and the linear decrease of the CD signal from 0.15 to 0.3 mg/mL in the 197 nm region. Therefore the Rpn values at 0.6 mg/mL are calculated as average value from the Rpn at 0.15 and 0.3 mg/mL collagen concentration.
Electrokinetic experiments
The Microslit Electrokinetic Setup, or MES—a dedicated instrument recently developed by the authors for the combined determination of streaming current/streaming potential and surface conductivity with planar samples (29)—was used to follow the charge formation process on collagen in aqueous solutions of varied type and concentration of electrolytes and pH. In brief, the experiment consists of streaming current measurements at various pressure differences across rectangular slit channels formed between two planar glass carriers coated with the collagen fibril layers (channel height 50 μm; see Fig. 1). Solutions for the electrokinetic measurements were prepared from vacuum-degassed MilliQ water by addition of 0.1 M potassium chloride, calcium chloride, potassium hydroxide, and hydrochloric acid solutions (VWR, Darmstadt, Germany). The measurements were performed at room temperature (23°C ± 1°C). Average values of three independent measurements are reported.
FIGURE 1.

Schematic drawing of the slit channel formed by two collagen-coated glass carriers for electrokinetic measurements.
RESULTS
DSC
Thermal denaturation of collagen samples was previously shown to cause the destruction of the triple helix and the unfolding of separated single peptide strains into a random coil (19,32). Thus, DSC experiments can provide information about the temperature Td and enthalpy ΔdH of the denaturation process; they do not allow us to distinguish contributions of secondary, tertiary, and quaternary structures.
A similar behavior of differently prepared samples (types A and B, see Materials and Methods) at a given electrolyte solution was observed. This may indicate that after sufficient time (four-day dialysis) no difference of the thermodynamic status persists between collagen samples prepared by the different procedures. The optical characteristics of the samples were found to be similar; optically clear in 10−4 M KCl and slightly turbid in 10−2 M KCl solutions. Also, the denaturation temperatures (i.e., Td for collagen in 10−4 M KCl solution, type A, 42.6 ± 0.4°C; type B, 42.8 ± 0.2°C) and enthalpies (i.e., ΔdH for collagen in 10−4 M KCl solution, type A, 46 ± 1 J/g; type B, 45 ± 1 J/g) solution match each other (see Fig. 2). Bearing in mind the similarity of samples prepared by either procedure, only results of experiments with samples prepared using procedure A will be reported and discussed below.
FIGURE 2.

DSC measurements: denaturation of collagen samples in 10−4 M KCl solutions prepared after two different sample preparation procedures (A, dialysis of the collagen solution and B, electrolyte PBS and heating to 37°C before dialysis; see Materials and Methods) with collagen concentration 0.6 mg/mL, heating rate 0.5 K/min.
The denaturation temperature (Td) of collagen samples was significantly increased from 42.6 ± 0.4°C to 47.2 ± 0.6°C with increasing ionic strength from 10−4 M to 10−2 M KCl. Along that line, an increased denaturation enthalpy from 46 ± 1 J/g to 60 ± 1 J/g was observed (see Fig. 3 and Table 2).
FIGURE 3.

DSC measurements: denaturation of collagen samples dialyzed against different KCl solutions, collagen concentration 0.6 mg/mL, heating rate 0.5 K/min.
TABLE 2.
Denaturation temperatures, enthalpies (DSC), and Rpn values (CD)
| DSC
|
||||
|---|---|---|---|---|
| I [M] | Td [°C] | ΔH [J/g] | ΔMH* [kJ/mol] | CD Rpn |
| 10−4 KCl | 42.6 ± 0.4 | 46 ± 1 | 13198 ± 239 | 0.13 ± 0.01 |
| 10−3 KCl | 44.0 ± 0.3 | 51 ± 1 | 14502 ± 389 | 0.16 ± 0.01 |
| 10−2 KCl | 47.2 ± 0.6 | 60 ± 1 | 17047 ± 309 | 0.43 ± 0.01 |
| 10−4 CaCl2 | 43.4 ± 0.2 | 48 ± 1 | 13817 ± 228 | 0.14 ± 0.01 |
| 10−3 CaCl2 | 43.9 ± 0.2 | 52 ± 1 | 14901 ± 285 | 0.16 ± 0.01 |
| 10−2 CaCl2 | 44.8 ± 0.1 | 55 ± 1 | 15738 ± 228 | 0.27 ± 0.01 |
Molar denaturation enthalpy, calculated with M = 285,000 g/mol, the collagen concentration was cColl = 0.6 mg/ml. The heating rate in the DSC experiments was 0.5 K/min.
In the case of the electrolyte CaCl2 the denaturation temperature (Td) of collagen samples increased slightly from 43.4 ± 0.2°C to 44.8 ± 0.3°C with increasing ionic strength from 10−4 M to 10−2 M CaCl2. Correspondingly a small increase of the denaturation enthalpy from 48 ± 1 J/g to 55 ± 1 J/g was observed (see Fig. 4 and Table 2). Compared to the results in the electrolyte system KCl, the collagen samples at the lowest CaCl2 solution concentration are only marginally more stable (see Table 2). At an ionic strength of 10−3 M the thermal stability of collagen in KCl and CaCl2 almost coincide, which manifests itself in both denaturation temperature and enthalpy (see Table 2). However, for samples in 10−2 M CaCl2 a lower thermal stability was observed than in 10−2 M KCl (lower denaturation temperature (44.8 ± 0.3°C vs. 47.2 ± 0.6°C in KCl) and a lower denaturation enthalpy (55 ± 1 J/g vs. 60 ± 1 J/g in KCl, see Table 2)).
FIGURE 4.

DSC measurements: denaturation of collagen samples dialyzed against different CaCl2 solutions, collagen concentration 0.6 mg/mL, heating rate 0.5 K/min.
CD
Circular dichroism is particularly sensitive to the secondary structure of proteins and therefore widely applied for conformational studies (33–35). However, CD-spectra of collagen samples differ from those of the vast majority of other proteins. Typically, collagen-like triple helical and poly-L-proline II like structures, respectively, show a large negative peak near 200 nm and a weaker positive peak at ∼220–225 nm, which is assigned to the triple helical structure (16,36–39). A very important parameter related to the peak intensities is the absolute value of the ratio of the dichroic intensity of the positive peak to the negative peak (Rpn), which can be considered as an index for triple helicity (39,40). During thermal denaturation the positive peak near 220–225 nm disappears and the intensity of the negative peak is decreased (36,39,41).
The ionic strength and the collagen concentration were systematically varied and the Rpn values were determined. The Rpn increased with ionic strength from 0.13 in 10−4 M KCl to 0.43 in 10−2 M KCl solution (Table 1). CD measurements were performed at collagen concentrations of 0.15, 0.3, and 0.6 mg/mL. Due to the strength of the UV signal at ∼200 nm in the case of 0.6 mg/mL collagen in 10−4 and 10−3 M KCl solutions, Rpn could be obtained reliably only for the more dilute samples containing 0.15 and 0.3 mg/mL collagen. Because of the linear increase of the CD signal in the 221-nm region (positive peak) for all collagen concentrations and the linear decrease of the CD signal from 0.15 to 0.3 mg/mL in the 197-nm region, the Rpn was assumed to be constant during measurement at different collagen concentrations at a given ionic strength of KCl. Therefore, the Rpn for 0.6 mg/mL was calculated as an average from the Rpn at 0.15 and 0.30 mg/mL (see Figs. 5 and 6, and Table 1). In 10−2 M KCl solution, Rpn values were found to be increased and the positions of the minimum (197 to 202 nm), the cross point (213 to 215 nm), and the maximum (221 to 223 nm) showed a minor shift toward higher wavelength when compared with samples in 10−4 M KCl solutions (see Fig. 7 and Table 1).
FIGURE 5.

CD spectra for different collagen concentrations dialyzed against 10−4 M KCl solution.
FIGURE 6.

CD spectra for different collagen concentrations dialyzed against 10−3 M KCl solution.
FIGURE 7.

CD spectra for different collagen concentrations dialyzed against 10−2 M KCl solution.
In CaCl2 solutions of collagen the Rpn increased with the ionic strength from 0.14 at 10−4 M CaCl2 to 0.27 at 10−2 M CaCl2 (see Table 1). In analogy to the results for the electrolyte system KCl, the Rpn was found to be constant upon increasing the collagen concentration at a given electrolyte solution with the same restrictions in the case of 0.6 mg/mL at 10−4 M CaCl2 and 10−3 M CaCl2 solutions discussed before. The Rpn for 10−4 M CaCl2 was slightly higher than for KCl (0.14 vs. 0.13) and the Rpn for 10−3 M CaCl2 approaches the value (0.16) for the corresponding KCl solution.
These results are in line with the DSC results (see Table 2). As might be expected given the lower denaturation enthalpy (see Table 2), the Rpn is smaller in 10−2 M CaCl2 than in the related KCl solution (0.27 compared to 0.43; see Table 2). In line with the increasing Rpn at 10−2 M CaCl2 solution the positions of the minimum (197–200 nm), the cross point (213–214 nm), and the maximum (221–222 nm) showed a minor shift toward higher wavelengths compared with the collagen samples dialyzed against 10−4 M / 10−3 M CaCl2 solutions, but the shift was smaller than the one observed for the KCl system.
Electrokinetic experiments
Surface-bound collagen I fibrils on glass carriers were prepared with high reproducibility as described in detail in Salchert et al. (30) for subsequent electrokinetic characterization in different solutions. The layered substrates have been thoroughly characterized with respect to immobilized amount and morphology (Figs. 8 and 9 give examples of images obtained by atomic force microscopy and scanning electron microscopy, respectively).
FIGURE 8.

Phase image of a collagen fibril layer in 10−2 M KCl solution obtained by atomic force microscopy.
FIGURE 9.

Electron microscopy picture of a dried collagen fibril layer, magnification 20,000×.
In the electrokinetic experiments streaming current versus pressure gradient data (dIS/dp) were determined as a function of pH in I = 10−4, 10−3, and 10−2 M KCl and CaCl2 solutions. Although streaming current and streaming potential data are often converted into electrokinetic (ζ) potentials, we do not report ζ-potential data for the collagen fibril layers since the assumptions underlying such a conversion are no longer justified when the structure of the layers (see Figs. 8 and 9) is irregular on a length scale of the extension of the electrical double layer. Nevertheless, the pH-dependent streaming current data provide information about the charge formation process of the collagen layer, which is determined by the varying ionization of amino-acid side chains of the protein. The charging of the collagen layers was found to be strongly influenced by the solution pH as well as by the type and the concentration of the “background” electrolyte.
The isoelectric point (IEP) was shifted significantly from pH 7.5 to 5.3 upon increase of the ionic strength from 10−4 M to 10−2 M KCl (see Fig. 10). This shift was found to be reversible and independent of the sample history. Since the IEP (and its shift with the ionic strength) was not influenced by the amount of the immobilized collagen (variation of the collagen surface concentration between 8.9 μg/cm2 and 26.3 μg/cm2 did not influence the results, data not shown), it was concluded that the collagen model layers used in this study reasonably represent the characteristics of collagen samples in the bulk phase.
FIGURE 10.

pH dependence of (dIS/dp) for the collagen fibril layer at different ionic strengths, electrolyte system KCl.
The pH dependence of the streaming current versus pressure gradient (dIS/dp) was also measured in 10−4, 10−3, and 10−2 M CaCl2 solutions (note that the ionic strength was comparable to the KCl measurements, the concentrations of CaCl2 were 3.3 × 10−5 M, 3.3 × 10−4 M, and 3.3 × 10−3 M). Again, the collagen layers showed a charging behavior with a strong dependence on the background electrolyte concentration. In contrast to the KCl electrolyte system the isoelectric point (IEP) shifted from pH 7.5 to pH 9 upon increase of the ionic strength from 10−4 M to 10−3 M CaCl2 (see Fig. 11). The IEP was not even reached within the accessible pH window at 10−2 M CaCl2 solution, indicating an even more basic IEP in case of the higher amount of CaCl2 (see Fig. 11).
FIGURE 11.

pH dependence of (dIS/dp) for the collagen fibril layer at different ionic strengths, electrolyte system CaCl2.
DISCUSSION
The results collected in our study point at the sensitive regulation of the ionization of collagen I by both the pH and electrolyte composition of the protein's aqueous environment. By means of the electrokinetic technique of streaming current measurements we were able for the first time to monitor the pH-dependent acid-base behavior of collagen I fibrils in situ using either KCl or CaCl2 as background electrolytes in a range of ionic strength between 10−4 and 10−2 M. A comparison with experiments characterizing the thermal stability (DSC) and conformation (CD) in similar solutions revealed a far-reaching correlation of the ionization of collagen I with the degree and stability of secondary structure of the protein.
A strong influence of the ionic strength on the charging behavior of the collagen I layers was found, as seen by the distinct downshift of the IEP with increasing KCl concentration (Fig. 10). A corresponding increase of ionic strength in the divalent electrolyte system CaCl2, on the other hand, shifts the IEP to more basic values (Fig. 11). Interestingly, our DSC results show an increase of the thermal stability with increasing ionic strength for both electrolyte systems (Figs. 3 and 4, and Table 2), which is expressed by the increase of denaturation temperature and the denaturation enthalpy. This finding is in line with the CD results, which show an increase of helicity in the same range (Table 2), indicating that the increase in thermal stability of the collagen molecules runs parallel with a change in the conformation of the protein. At low ionic strengths (up to 10−3 M) there is no difference between the KCl and the CaCl2 electrolyte system, but at higher ionic strengths CaCl2 proves less efficient in stabilizing the protein than KCl (Table 2). These results suggest that the thermal and conformational stability of collagen I is strongly influenced by electrostatic interactions, which were analyzed in situ for the first time by means of electrokinetic experiments complementing the classic approach of DSC and CD techniques.
The role of electrostatic interactions for the stability of proteins has been discussed controversially by different authors (42–50). It is generally accepted that there are important nonspecific interactions like hydrophobic and van der Waals interactions, where hydrophobic interactions are the major driving force for protein folding. Specific interactions, which are mostly electrostatic, are important for protein flexibility, function, and folding (42). Theoretical calculations comparing the interaction of charged residues to interactions between hydrophobic residues of similar size and shape suggested that close-range electrostatic interactions rather destabilize a protein (43–45). More recently theoretical (46–48) and experimental studies (49,50) proposed that the majority of salt bridges are stabilizing, but further indicated that the individual geometrical orientation of the partners in any salt bridge plays an important role for the stability of the bridge. According to Kumar and Nussinov (42) the contribution of one salt bridge can be subdivided into three terms: Term I, favorable bridging energy due to the direct interaction of oppositely charged side chains; Term II, desolvation penalties for the individual bridging residue (due to the change of the structure of the environment from water to protein); and Term III, the interaction of the salt-bridging side chains with the charges in the rest of the protein. Furthermore the authors discuss the geometrical orientation as a major criterion for the salt-bridge stability and categorize ion pairs into most favorable salt bridges, less favorable N-O bridges, and most unfavorable longer-range ion pairs. Following the authors, an ion pair is defined as a favorable salt bridge “if the centroids of the side chain charged group atoms in the residues lie within 4.0 Å of each other and at least one pair of Asp or Glu side-chain carbonyl oxygen and side chain nitrogen atoms of Arg, Lys, or His are also within this distance.” Accomplishing the second criteria but missing the first one (centroids are not in 4 Å distance) leads to a less favorable N-O bridge and missing both criteria implies a long-range ion pair which is mostly destabilizing. A specific limitation of collagen in creating intra- and interchain interactions is caused by the linear nature of the triple helix. Molecular modeling of triple helical sequences (51) showed possible interchain interactions between the Y (or X′) residue in one chain and the X (or Y) residue in the adjacent chain (staggered by one), writing neighboring triplets as Gly-X-Y-Gly-X′-Y′-Gly-X″-Y″. Furthermore intrachain interactions of X-X′ and Y-Y′ residues are possible, but interactions of side chains separated by more than three residues are not possible (no X-Y′ or Y-X″ (17,51)). This is consistent with the criteria of Kumar and Nussinov (42), indicating that only closely neighboring residues are able to form stabilizing interactions. These considerations clearly imply an important role of the electrolyte composition on the efficacy of salt bridges as the screening of charged moieties and the range of electrostatic interactions is massively influenced by the ionic strength of the solution.
As described before (see Introduction) the range of electrostatic interactions (Debye screening length κ−1) is substantially reduced from 30 nm to 3 nm in the investigated range of ionic strengths (from 10−4 to 10−2 M, see Fig. 12). Following the results at lower ionic strength the destabilization of the triple helix structure could be caused by long-range electrostatic interactions, where mostly unfavorable longer-range ion pairs could occur (42). The possibility for one single charged side chain to interact with just one other single charged residue to form a favorable salt bridge is nearly impossible under such long-range (κ−1 = 30 nm) conditions. Instead, each charged residue will experience a rather diffuse electrostatic interaction with a variety of other residues (see Fig. 12, top).
FIGURE 12.

Schematic view of the range of electrostatic interactions at different ionic strengths: (top) I = 10−4 M, very long range of electrostatic interactions (Debye length 30 nm) and (bottom) I = 10−2 M, shorter range of electrostatic interactions (Debye length 3 nm).
This picture is consistent with theoretical insights about the interaction of heterogeneously charged infinite rods. The screened electric potential of a model polyelectrolyte consisting of a linear array of evenly spaced point charges q along the z axis with intercharge spacing d is given, according to Debye-Hückel theory, by (53)
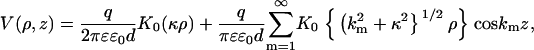 |
(3) |
with km = 2πm/d, (m = 1,2,3..). Here, (ρ, z) are cylindrical coordinates and K0(x) is the modified Bessel function with the asymptotic behavior 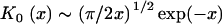 for large x. The first term on the right-hand side of Eq. 3 describes the potential due to the homogeneously smeared-out mean charge of the model polyelectrolyte. This type of contribution is responsible for an electrostatic repulsion between two polyelectrolytes of equal overall charge; it only depends on the radial coordinate ρ and for large separation decays with a characteristic decay length given by the Debye screening length κ−1. The terms in the sum on the right describe modulations in the potential arising from the discrete distribution of charges along the model polyelectrolyte. This charge heterogeneity contributes an attractive interaction between similar polyelectrolytes at short separations. The radial range of the potential modulations is
for large x. The first term on the right-hand side of Eq. 3 describes the potential due to the homogeneously smeared-out mean charge of the model polyelectrolyte. This type of contribution is responsible for an electrostatic repulsion between two polyelectrolytes of equal overall charge; it only depends on the radial coordinate ρ and for large separation decays with a characteristic decay length given by the Debye screening length κ−1. The terms in the sum on the right describe modulations in the potential arising from the discrete distribution of charges along the model polyelectrolyte. This charge heterogeneity contributes an attractive interaction between similar polyelectrolytes at short separations. The radial range of the potential modulations is 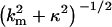 , which implies that modulations caused by charge fluctuations on a length scale shorter than the Debye length decay much faster than the homogeneous potential contributed by the mean charge density. If the Debye length becomes much larger than the characteristic length of the fluctuations, then the polyelectrolyte's charge heterogeneity becomes virtually invisible to any probe charge. On similar grounds, one might expect that a charged group on a collagen chain will not feel the charge heterogeneity given by distribution of ionizable groups on a second collagen chain and therefore cannot form salt bridges when the Debye length is large.
, which implies that modulations caused by charge fluctuations on a length scale shorter than the Debye length decay much faster than the homogeneous potential contributed by the mean charge density. If the Debye length becomes much larger than the characteristic length of the fluctuations, then the polyelectrolyte's charge heterogeneity becomes virtually invisible to any probe charge. On similar grounds, one might expect that a charged group on a collagen chain will not feel the charge heterogeneity given by distribution of ionizable groups on a second collagen chain and therefore cannot form salt bridges when the Debye length is large.
In line with this, the charging of the collagen fibril layer appears to be determined by the superposition of the acid-base behavior of all ionizable functionalities of the collagen molecules. Assuming that long-range electrostatic interactions reduce the number of salt bridges as discussed above, nearly all amino-acid residues should contribute to the electrical charging of the protein at low ionic strength. Therefore the IEP of 7.5 in both electrolyte systems correlates well with the amino-acid sequence of the collagen I molecule, which shows a slight excess of positively charged residues. However, increasing the ionic strength in the KCl system leads to more acidic net charge characteristics, accompanied by an increased thermal and conformational stability of the collagen. The observed increase of stability and helicity in the collagen molecule at elevated ionic strengths can be explained with the formation of a higher number of favorable salt bridges, which would also lead to a significant reduction of the density of free charged residues on the molecule. Formation of salt bridges, however, cannot explain the negative net charge observed down to a pH of 5.3 in 10−2 M KCl solutions, since salt bridge formation always involves two oppositely charged amino-acid side chains and thus conserves the net charge.
A shift of the pKeff of ionized functionalities—as discussed for several proteins (54,55)—was considered to explain the observed variation of the IEP with altered ionic strength. However, for the particular case of the collagen molecule this explanation was found to be unlikely. First of all, the amino-acid sequence shows a slight excess of positively charged groups, but the IEP shifts with increasing ionic strength to a lower pH-region (more acidic). This behavior requires a massive downshift of 3–4 pH units for the usually high pKint values of lysine (≈10.4, (56,57)) and arginine-residues (≈12.0, (56,57)) together with an unaltered or downshifted pKeff of the acidic groups. There are no reports of such a strong shift of the pKeff values for lysine and arginine; instead pKeff values at ∼10.5 are reported for lysine residues in different proteins (58). Solvent-accessibility calculations based on the algorithm of Lee and Richards (13) point at a rather high accessibility of all types of ionizable residues in the collagen molecule: For the characteristic collagen sequence of Gly-X-Y triplets, average solvent-accessibility factors of ionizable residues were found to be 0.77 at the X-position and 0.63 for the Y-position, where 0.01 corresponds to complete burial and 1.0 to full exposure. Therefore, extreme shifts of the pKeff due to partial burial of lysine and arginine groups in the rodlike collagen molecule can be excluded. This was corroborated by calculations using the solvent accessibility-modified (59) Tanford-Roxby (60) iterative approximation to estimate the electrostatic interaction potential for charged sites according to the Debye-Hückel theory (53,61). To consider the maximum shift, only positive charges (identical charges) with equal spacing of 0.286 nm (close contact of neighboring residues (5)) on a cylinder with a diameter of 1.5 nm (5) and a dielectric constant of 3 were assumed. The results for an intrinsic pKint of 10.4 of the lysine groups indicate only a minor shift of ΔpH = 0.01 with altered ionic strength (pKeff 10.26 in 10−4 M KCl and 10.27 in 10−2 M KCl). These findings rule out pK-shifts as the origin of the IEP's distinct downshift with increasing ionic strength.
Lowering the density of charged groups on the collagen by the reduced screening length (see Fig. 12, bottom) and the formation of stabilizing salt bridges at higher ionic strength may, however, lead to an increasing influence of asymmetrical ion adsorption on the net charge formation. Although preferential adsorption of chloride ions was discussed in some of the earlier studies (26,62), there is evidence for the indifferent adsorption behavior of chloride and potassium on collagen (63,64). It is, however, well known from studies on a variety of polymeric materials that preferential adsorption of hydroxide ions leads to a net negative charge of surfaces without any ionizable surface groups for pH values above pH 4 (29,65,66). A similar adsorption mechanism could be expected to provide charge to nondissociating amino-acid side chains, especially at high ionic strength, where interference from the charge of nearby dissociating sites is reduced by strong screening.
In consequence, a stronger contribution of uncharged domains could affect the charge formation process at higher ionic strength, causing the negative net charge of collagen molecules at higher ionic strengths in the electrolyte system KCl.
Following the electrokinetic results at lower ionic strength, the destabilization of the triple helix structure could be caused by long-range electrostatic interactions, where mostly unfavorable longer-range ion pairs could occur (42). Therefore the helicity is reduced with respect to the more native conditions at higher ionic strengths. Conversely, reducing the range of electrostatic interactions (see Fig. 12, bottom) by increasing the ionic strength could lead to the formation of more favorable salt bridges, induced by better matching of oppositely charged side chains (in analogy to Term I of (42) discussed above), accompanied by an altered and reduced interaction of the salt-bridging side chains with the charges in the rest of the triple helix (analogously to Term III (42)). Next, a reduction of unfavorable “long-range electrostatic interactions” itself could increase the conformational stability by decreasing the number of charged residues not involved in salt bridges and allowing for stronger nonspecific hydrophobic and van der Waals interactions. These phenomena could be the reasons for the increased thermal stability and helicity observed at higher ionic strength.
Given the observed changes in enthalpy and helicity (see DSC and CD, Table 2) and the lower IEP at higher ionic strengths, a high amount of favorable ion pairs should occur in the related more native triple helices. In fact, Persikov et al. (17,51) found that Lys-Gly-Asp/Glu sequences in host guest peptides are highly stabilizing by forming interchain salt bridges. Analysis of sequence data (67) showed a frequent amount of Lys-Gly-Asp/Glu triplets in collagen I (e.g., 12 Lys-Gly-Asp and 22 Lys-Gly-Glu triplets in human collagen I) supporting this assumption. Furthermore, substantial electrostatic stabilization by intrachain salt bridges were observed by charged residues in Y-Y′ positions (17) and sequence analysis with consideration of molecular structure shows that 60% of the charged side chains are able to create salt bridges in collagen I (13).
The charge formation process in calcium chloride solutions seems to be determined by preferential interactions of the divalent calcium ions with the negatively charged amino-acid side chains (64): The calcium ion contains two charges in a rather small volume (radius of 4.1 Å in the hydrated state (68)), and is therefore surrounded by a strong electric field that favors interactions with the negatively charged Asp or Glu side chains. Geometrical restrictions of the accessibility of charged side chains by calcium ions can be excluded, as the radius of 4.1 Å for hydrated calcium ions fits well to the distance of ∼14 Å between adjacent triple helical molecules in collagen like proteins and collagen in tissues determined by fiber diffraction (69). At low ionic strength the long-range electrostatic interaction causes destabilization of collagen as described for the KCl system (seen in nearly similar charging behavior, thermal, and conformational stability). Increasing the ionic strength with CaCl2 first of all leads to a stabilization (see Table 2), which is consistent with the KCl system up to 10−3 M and probably related to the same effects of reducing the range of electrostatic interactions. Further increase of the CaCl2 concentration still reduces the range of electrostatic forces (resulting in increased stability from I = 10−3 to 10−2 M CaCl2, see Table 2), but simultaneously leads to the overcompensation of negatively charged residues, as is obvious from the shift of the IEP to the basic pH range (Fig. 11). This overcompensation by calcium ions destabilizes the collagen molecules: negatively charged Glu and Asp residues acquire a net charge of +1 through the Ca2+-binding and thus experience an electrostatic repulsion from the positively charged Lys and Arg side chains.
Altogether increasing the ionic strength by addition of CaCl2 leads to a positively charged collagen molecule that is less stable than the protein at similar ionic strength in the simple 1:1 electrolyte system KCl.
CONCLUSIONS
The acid base behavior of collagen I was found to significantly depend on the ionic strength in KCl as well as in CaCl2 solutions. An increase of the ionic strength in the KCl system causes a more acidic (negatively charged) collagen molecule where an increase of the ionic strength in the CaCl2-system leads to a more basic (positively charged) collagen molecule.
The shift of the protein's IEP to higher values at elevated concentrations of calcium ions can be explained by the binding of divalent positive calcium ions to the carboxylic acid anions of the glutamic- and aspartic-acid side chains. Beyond that, the charge formation process in KCl solutions was found to be determined by the ionizable functionalities of the protein at low ionic strength but could be additionally influenced by preferential adsorption of hydroxide ions onto uncharged surface sites at higher ionic strength due to the formation of stabilizing short-ranging salt bridges and the lower Debye screening length.
An increase of the thermal stability with increasing ionic strength was observed for both electrolyte systems by DSC analysis. This finding is in line with the CD results, which show an increase of helicity over the same range of electrolyte concentrations, indicating that the increase in thermal stability of the collagen molecules runs parallel with a change in the conformation of the protein.
Large screening (Debye) lengths, which occur at low ionic strength, favor the formation of longer-range ion pairs, which are mostly destabilizing and therefore could explain the lower conformational stability of the collagen molecule at these conditions. The formation of stabilizing short-ranging ion pairs is promoted at low Debye lengths, which agrees well with the increased thermal stability and helicity observed at higher ionic strengths. While divalent calcium ions stabilize the protein at lower ionic strengths (up to 10−3 M) in line with this explanation, the overcompensation of the charge of anionic amino-acid side chains at higher CaCl2 concentrations was found to cause an additional destabilizing contribution.
Acknowledgments
Contributions of T. Osaki, R. Zimmermann, C. Rauwolf, and A. Schöne for helpful discussions and T. Pompe for atomic force measurements (all Leibniz Institute of Polymer Research Dresden) are gratefully acknowledged.
This work was financed by the BASF AG, Ludwigshafen.
Sven H. Behrens's current address is Georgia Institute of Technology, School of Chemical & Biomolecular Engineering, Atlanta, GA 30332-0100.
References
- 1.Lutolf, M. P., and J. A. Hubbell. 2005. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 23:47–55. [DOI] [PubMed] [Google Scholar]
- 2.Reich, G. 1995. Collagen report: a review about the present state. Leder. 46:192–199. [Google Scholar]
- 3.Bayley, A. J., and R. G. Paul. 1998. Collagen: a not so simple protein. J. Soc. Leather Technol. Chem. 83:104–110. [Google Scholar]
- 4.Cot, J. 2004. An imaginary journey to the collagen molecule for a better understanding of leather waste treatments. J. Am. Leather Chem. Assoc. 99:322–350. [Google Scholar]
- 5.Engel, J., and H. J. Bächinger. 2005. Structure, stability and folding of the collagen triple helix. Top. Curr. Chem. 247:7–33. [Google Scholar]
- 6.Rich, A., and F. H. Crick. 1961. The molecular structure of collagen. J. Mol. Biol. 3:483–506. [DOI] [PubMed] [Google Scholar]
- 7.Ramachandran, G. N., editor. 1964. Treatise on Collagen. Academic Press, New York.
- 8.Bella, J., M. Eaton, B. Brodsky, and H. M. Berman. 1994. Crystal and molecular structure of a collagen-like peptide at 1.9 Å resolution. Science. 266:75–81. [DOI] [PubMed] [Google Scholar]
- 9.Kramer, R. Z., J. Bella, P. Mayville, B. Brodsky, and H. M. Berman. 1999. Sequence dependent conformational variations of collagen triple-helical structure. Nat. Struct. Biol. 6:454–457. [DOI] [PubMed] [Google Scholar]
- 10.Hansen, U., and P. Bruckner. 2003. Macromolecular specificity of collagen fibrillogenesis: fibrils of collagens I and XI contain a heterotypic alloyed core and a collagen I sheath. J. Biol. Chem. 278:37352–37359. [DOI] [PubMed] [Google Scholar]
- 11.Steplewski, A., H. Ito, E. Rucker, R. J. Brittingham, T. Alabyeva, M. Gandhi, F. K. Ko, D. E. Birk, S. A Jimenez, and A. Fertala. 2004. Position of single amino acid substitutions in the collagen triple helix determines their effect on structure of collagen fibrils. J. Struct. Biol. 148:326–337. [DOI] [PubMed] [Google Scholar]
- 12.Wenstrup, R. J., J. B. Florer, E. W. Brunskill, S. M. Bell, I. Chervoneva, and D. E. Birk. 2004. Type V collagen controls the initiation of collagen fibril assembly. J. Biol. Chem. 279:53331–53337. [DOI] [PubMed] [Google Scholar]
- 13.Jones, E. Y., and A. J. Miller. 1991. Analysis of structural design features in collagen. J. Mol. Biol. 218:209–219. [DOI] [PubMed] [Google Scholar]
- 14.Chan, V. C., J. A. M. Ramshaw, A. Kirkpatrick, K. Beck, and B. Brodsky. 1997. Positional preferences of ionizable residues in Gly-X-Y triplets of the collagen triple-helix. J. Biol. Chem. 272:31441–31446. [DOI] [PubMed] [Google Scholar]
- 15.Singh, M. P., J. Stefko, J. A. Lumpkin, and J. Rosenblatt. 1995. The effect of electrostatic charge interactions on release rates of gentamicin from collagen matrices. Pharm. Res. 12:1205–1210. [DOI] [PubMed] [Google Scholar]
- 16.Venugopal, M. G., J. A. M. Ramshaw, E. Braswell, D. Zhu, and B. Brodsky. 1994. Electrostatic Interactions in collagen-like triple-helical peptides. Biochemistry. 33:7948–7956. [DOI] [PubMed] [Google Scholar]
- 17.Persikov, A. V., J. A. M. Ramshaw, A. Kirkpatrick, and B. Brodsky. 2005. Electrostatic interactions involving lysine make major contributions to collagen triple-helix stability. Biochemistry. 44:1414–1422. [DOI] [PubMed] [Google Scholar]
- 18.Katz, E. P., and C. W. David. 1990. Energetics of intrachain salt-linkage formation in collagen. Biopolymers. 29:791–798. [DOI] [PubMed] [Google Scholar]
- 19.Friess, W., and G. Lee. 1996. Basic thermoanalytical studies of insoluble collagen matrices. Biomaterials. 17:2289–2294. [DOI] [PubMed] [Google Scholar]
- 20.Miles, C. A., and A. J. Bailey. 2001. Thermally labile domains in the collagen molecule. Micron. 32:325–332. [DOI] [PubMed] [Google Scholar]
- 21.Leikina, E., M. V. Mertts, N. Kuznetsova, and S. Leikin. 2002. Type I collagen is thermally unstable at body temperature. Proc. Natl. Acad. Sci. USA. 99:1314–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bianchi, E., and G. Conio. 1967. The role of pH, Temperature, salt type, and salt concentration on the stability of the crystalline, helical and randomly coiled forms of collagen. J. Biol. Chem. 242:1361–1369. [PubMed] [Google Scholar]
- 23.Miles, C. A., T. V. Burjanadze, and A. J. Bailey. 1995. The kinetics of the thermal denaturation of collagen in unrestrained rat tail tendon determined by differential scanning calorimetry. J. Mol. Biol. 245:437–446. [DOI] [PubMed] [Google Scholar]
- 24.Tiktopulo, E. I., and A. V. Kajava. 1998. Denaturation of type I collagen fibrils is an endothermic process accompanied by a noticeable change in the partial heat capacity. Biochemistry. 37:8147–8152. [DOI] [PubMed] [Google Scholar]
- 25.Usha, R., and T. Ramasami. 2000. Effect of pH on dimensional stability of rat tail tendon collagen fiber. J. Appl. Polym. Sci. 75:1577–1584. [Google Scholar]
- 26.Aktas, N. 2003. The effects of pH, NaCl and CaCl2 on the thermal denaturation characteristics of intramuscular connective tissue. Thermochim. Acta. 407:105–112. [Google Scholar]
- 27.Brown, E. M., H. M. Farrell, and R. J. Wildermuth. 2000. Influence of neutral salts on the hydrothermal stability of acid-soluble collagen. J. Protein Chem. 19:85–92. [DOI] [PubMed] [Google Scholar]
- 28.Komsa-Penkova, R., R. Koynova, G. Kostov, and B. G. Tenchov. 1996. Thermal stability of calf skin collagen type I in salt solutions. Biochim. Biophys. Acta. 1297:171–181. [DOI] [PubMed] [Google Scholar]
- 29.Werner, C., H. Körber, R. Zimmermann, S. Dukhin, and H.-J. Jacobasch. 1998. Extended electrokinetic characterization of flat solid surfaces. J. Colloid Interface Sci. 208:329–346. [DOI] [PubMed] [Google Scholar]
- 30.Salchert, K., U. Streller, T. Pompe, N. Herold, M. Grimmer, and C. Werner. 2004. In vitro reconstitution of fibrillar collagen type I assemblies at reactive polymer surfaces. Biomacromolecules. 5:1340–1350. [DOI] [PubMed] [Google Scholar]
- 31.Pompe, T., S. Zschoche, N. Herold, K. Salchert, M.-F. Gouzy, C. Sperling, and C. Werner. 2003. Maleic anhydride copolymers—a versatile platform for molecular biosurface engineering. Biomacromolecules. 4:1072–1079. [DOI] [PubMed] [Google Scholar]
- 32.Payne, K. J., and A. Veis. 1988. Fourier transform IR spectroscopy of collagen and gelatin solutions: deconvolution of the amide I band for conformational studies. Biopolymers. 27:1749–1760. [DOI] [PubMed] [Google Scholar]
- 33.Sreerama, N., and R. W. Woody. 1994. Protein secondary structure from circular dichroism spectroscopy. J. Mol. Biol. 242:497–507. [DOI] [PubMed] [Google Scholar]
- 34.Sreerama, N., S. Y. Venyaminov, and R. W. Woody. 1999. Estimation of the number of α-helical and β-strand segments in proteins using circular dichroism spectroscopy. Protein Sci. 8:370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sreerama, N., S. Y. Venyaminov, and R. W. Woody. 2000. Estimation of protein secondary structure from circular dichroism spectra: inclusion of denatured proteins with native proteins in the analysis. Anal. Biochem. 287:243–251. [DOI] [PubMed] [Google Scholar]
- 36.Tiffany, M. L., and S. Krimm. 1972. Effect of temperature on the circular dichroism spectra of polypeptides in the extended state. Biopolymers. 11:2309–2316. [DOI] [PubMed] [Google Scholar]
- 37.Sreerama, N., and R. W. Woody. 1994. Poly(pro)II helices in globular proteins: identification and circular dichroic analysis. Biochemistry. 33:10022–10025. [DOI] [PubMed] [Google Scholar]
- 38.Toumadje, A., and W. C. Johnson, Jr. 1995. Systemin has the characteristics of a poly(L-proline) II type helix. J. Am. Chem. Soc. 117:7023–7024. [Google Scholar]
- 39.Feng, Y., G. Melacini, J. P. Taulane, and M. Goodman. 1996. Acetyl-terminated and template-assembled collagen-based polypeptides composed of Gly-Pro-Hyp sequences. 2. Synthesis and conformational analysis by circular dichroism, ultraviolet absorbance, and optical rotation. J. Am. Chem. Soc. 118:10351–10358. [Google Scholar]
- 40.Barth, D., A. G. Milbradt, C. Renner, and L. Moroder. 2004. A(4R)- or a (4S)-fluoroproline residue in position XAA of the (XAA-YAA-Gly) collagen repeat severely affects triple helix formation. ChemBioChem. 5:79–86. [DOI] [PubMed] [Google Scholar]
- 41.Consonni, R., L. Zetta, R. Longhi, L. Toma, G. Zanaboni, and R. Tenni. 2000. Conformational analysis and stability of collagen peptides by CD and by 1H- and 13C- NMR spectroscopy. Biopolymers. 53:99–111. [DOI] [PubMed] [Google Scholar]
- 42.Kumar, S., and R. Nussinov. 2002. Close-range electrostatic interactions in proteins. ChemBioChem. 3:604–617. [DOI] [PubMed] [Google Scholar]
- 43.Hendsch, Z. S., and B. Tidor. 1994. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 3:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Honig, B., and A. Nicholls. 1995. Classical electrostatics in biology and chemistry. Science. 268:1144–1149. [DOI] [PubMed] [Google Scholar]
- 45.Xiao, L., and B. Honig. 1999. Electrostatic contributions to the stability of hyperthermophilic proteins. J. Mol. Biol. 289:1435–1444. [DOI] [PubMed] [Google Scholar]
- 46.Kumar, S., and R. Nussinov. 2001. Fluctuations in ion pairs and their stabilities in proteins. Proteins Struct. Funct. Genet. 43:433–454. [DOI] [PubMed] [Google Scholar]
- 47.Kumar, S., and R. Nussinov. 2002. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys. J. 83:1595–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong, F., and H. X. Zhou. 2002. Electrostatic contributions to T4 lysozyme stability: solvent-exposed charges versus semi-buried salt bridges. Biophys. J. 83:1341–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Makhatadze, G. I., V. V. Loladze, D. N. Ermolenko, X. Chen, and S. T. Thomas. 2003. Contribution of surface salt-bridges to protein stability: guidelines for protein engineering. J. Mol. Biol. 327:1135–1148. [DOI] [PubMed] [Google Scholar]
- 50.Molero, B. I., J. A. Zitzewitz, and C. R. Matthews. 2004. Salt-bridges can stabilize but not accelerate the folding of the homodimeric coiled-coil peptide GCN4-p1. J. Mol. Biol. 336:989–996. [DOI] [PubMed] [Google Scholar]
- 51.Persikov, A. V., J. A. M. Ramshaw, and B. Brodsky. 2005. Prediction of collagen stability from amino acid sequence. J. Biol. Chem. 280:19343–19349. [DOI] [PubMed] [Google Scholar]
- 52.Katz, E. P., and C. W. David. 1992. Unique side-chain conformation encoding for chirality and azimuthal orientation in the molecular packing of skin collagen. J. Mol. Biol. 228:963–969. [DOI] [PubMed] [Google Scholar]
- 53.Soumpasis, D. 1978. Debye-Hückel theory of model polyelectrolytes. J. Chem. Phys. 69:3190–3196. [Google Scholar]
- 54.Russell, A. J., and A. R. Fersht. 1987. Rational modification of enzyme catalysis by engineering surface charge. Nature. 328:496–500. [DOI] [PubMed] [Google Scholar]
- 55.Matthew, J. B., M. A. Flanagan, E. B. Garcia-Moreno, K. L. March, S. J. Shire, and F. R. N. Gurd. 1985. pH-dependent processes in proteins. CRC Crit. Rev. Biochem. 18:91–197. [DOI] [PubMed] [Google Scholar]
- 56.Matthew, J. B., and F. M. Richards. 1982. Anion binding and pH-dependent electrostatic effects in ribonuclease. Biochemistry. 21:4989–4999. [DOI] [PubMed] [Google Scholar]
- 57.Neves-Petersen, M. T., and S. B. Petersen. 2003. Protein electrostatics: a review of the equations and methods used to model electrostatic equations in biomolecules—applications in biotechnology. In Biotechnology Annual Review 9. M. R. El-Gewely, editor. Elsevier/North Holland, Amsterdam. [DOI] [PubMed]
- 58.Yang, A.-S., and B. Honig. 1993. On the pH dependence of protein stability. J. Mol. Biol. 231:459–474. [DOI] [PubMed] [Google Scholar]
- 59.Shire, S. J., I. H. Hanania, and F. R. N. Gurd. 1974. Electrostatic effects in myoglobins. Hydrogen ion equilibria in sperm whale ferrimyoglobin. Biochemistry. 13:2967–2974. [DOI] [PubMed] [Google Scholar]
- 60.Tanford, C., and R. Roxby. 1972. Interpretation of protein titration curves. Application to lysozyme. Biochemistry. 11:2192–2198. [DOI] [PubMed] [Google Scholar]
- 61.Borkovec, M., B. Jönsson, and G. J. M. Koper. 2001. Ionization processes and proton binding in polyprotic systems: small molecules, proteins, interfaces and polyelectrolytes. Surf. Colloid Sci. 16:99–339. [Google Scholar]
- 62.Jackson, D. S., and A. Neuberger. 1957. Observations on the isoionic and isoelectric point of acid-processed gelatin from insoluble and citrate-extracted collagen. Biochim. Biophys. Acta. 26:638–639. [DOI] [PubMed] [Google Scholar]
- 63.Gilbert, I. G. F. 1960. Microelectrophoretic studies of soluble collagen. Biochim. Biophys. Acta. 4:156–162. [DOI] [PubMed] [Google Scholar]
- 64.Li, S.-T., and E. P. Katz. 1976. An electrostatic model for collagen fibrils. The interaction of reconstituted collagen with Ca++, Na+, and Cl−. Biopolymers. 15:1439–1460. [DOI] [PubMed] [Google Scholar]
- 65.Zimmermann, R., S. Dukhin, and C. Werner. 2001. Electrokinetic measurements reveal interfacial charge at polymer films caused by simple electrolyte ions. J. Phys. Chem. B. 105:8544–8549. [Google Scholar]
- 66.Werner, C., U. König, A. Augsburg, C. Arnhold, H. Körber, R. Zimmermann, and H.-J. Jacobasch. 1999. Electrokinetic surface characterization of biomedical polymers—a survey. Colloid Surf. A. 159:519–529. [Google Scholar]
- 67.ExPASy Proteomics Server: Swiss-Prot/TrEMBL: P02452 (CO1A1_HUMAN), P08123 (CO1A2_HUMAN).
- 68.Israelachvili, J. N. 1992. Intermolecular and Surface Forces, 2nd Ed. Academic Press, London.
- 69.Brodsky, B., and J. A. M. Ramshaw. 1997. The collagen triple–helix structure. Matrix Biol. 15:545–554. [DOI] [PubMed] [Google Scholar]