

Abstract
A quantitative understanding of nucleic acid hybridization is essential to many aspects of biotechnology, such as DNA microarrays, as well as to the structure and folding kinetics of RNA. However, predictions of nucleic acid secondary structures have long been impeded by the presence of helices interior to loops, so-called pseudoknots, which impose complex three-dimensional conformational constraints. In this paper we compute the pseudoknot free energies analytically in terms of known standard parameters, and we show how the results can be included in a kinetic Monte Carlo code to follow the succession of secondary structures during quenched or sequential folding. For the hepatitis delta virus ribozyme, we predict several nonnative stems on the folding path, characterize a kinetically trapped state, interpret several experimentally characterized mutations in terms of the folding path, and suggest how hybridization with other parts of the genome inactivates the newly formed ribozyme.
The secondary structure elements of RNA, the base-paired helices, are energetically stable and thus building blocks for the three-dimensional configuration (1). Beyond the nested secondary structures whose minimum free energy configuration can be readily evaluated by dynamic programming (2) are those that contain stems interior to loops—i.e., the so-called pseudoknots. The prediction of these nonnested configurations by means of an exhaustive search algorithm is both difficult and computationally expensive (sequences currently limited to ≈130 nt) (3). By contrast, algorithms that seek to follow the actual folding dynamics (4) can in principle treat pseudoknotted and nested structures on similar bases.
RNA folding kinetics is known to proceed stochastically via a succession of partially folded secondary structures in quasi-equilibrium. Elementary transitions which make or break single helices can thus be assigned Arrhenius-like rates (5, 6), k = k° × exp(−ΔG/kT), where k° reflects only local stacking processes within a transient nucleation core. Hence, merely evaluating free energy differences ΔG between the transition states and the current configurations can provide access to the molecular folding dynamics. Within this so-called “kinetic Monte Carlo” scheme (4), the simulation actually follows the folding kinetics, in addition to providing the equilibrium distribution of states in the long time limit.
In Theory and Methods, we first show how the free energy contribution of pseudoknots can be evaluated by using polymer theory and known stacking free energies for the helices. We then outline the main steps of the kinetic Monte Carlo scheme for RNA. This includes, in particular, an efficient optimization scheme to account for the presence of competing helices in most partially folded as well as transition configurations along the folding path.
The first structure predictions are encouraging, as judged by experiments on small pseudoknotted RNAs, and constitute a base line against which more elaborate schemes that employ pseudoknot-specific parameters can be compared.
As for folding kinetics, the hepatitis delta virus (HDV) ribozyme is perhaps the best example on which to test our code. The ribozyme is active in vitro, and it has been the subject of many experiments that explore the effects of various mutations (7). Concentrating on its folding paths, we identify (i) a transient kinetic trap that dominates quenched folding and (ii) a succession of transient stems that cooperatively guide sequential folding. Our quantitative predictions, which could be directly tested experimentally (8, 9), largely support the suggestion that sequential folding kinetics of the HDV ribozyme directly regulate its in vivo function (7, 10), a feature also suspected for other ribozymes (1, 11).
Theory and Methods
Evaluating Free Energies.
We calculate a free energy G for an arbitrary secondary structure in terms of the thermodynamic free energies of the individual helices (12, 13) and a conformational entropy of the entire structure, including pseudoknots. All helices with 3 or more base pairs and a stem free energy of at least −10 kT (i.e., −6 kcal/mol) are included. Structurally, the helices are modeled as rods of appropriate length and the unpaired regions as Gaussian chains with Kuhn length b = 1.5 nm (14) or 2.5 bases of size a ≃ 6 Å. Conformational entropies are then evaluated in two stages, by first defining substructures we call nets (see Fig. 1 for details), whose entropy can be calculated exactly and analytically, and then treating constraints among nets more globally, by adopting a coarse-grained crosslinked-gel-like description of the RNA molecule at larger scales (see Fig. 2 for details). The introduction of these two complementary descriptions, appropriate for short and large scales, respectively, enables us to overcome the general “pseudoknot problem” with an algorithm that evaluates free energies of nested and nonnested secondary structures in about the same computation time.
Figure 1.

Short-scale description: The 8 allowed “nets.” We define “closed-nets” on a configuration by labeling all single-stranded closed circuits that can be drawn on the structure by hopping between adjacent single-stranded sections each time a stem end is encountered (see “closed-net-1”). (The dashed stems connect to other nets, but do not affect the order of the nets drawn. Other net-connecting stems may also be present along any single strand.) This procedure readily identifies hairpin, bulge, internal, and multibranched loops as other instances of closed-nets-0. However, the same procedure delimits also more complex single-stranded circuits (closed-nets-n) which pass through both ends of a number n of stems (i.e., n internal stems in a closed-net-n). Those nets correspond to locally pseudoknotted substructures. Similarly, for noncircular RNA sequences, we also partition the 5′ to 3′ open path into adjacent “open-nets-n” which are the continuous sections of the path that contain a minimal number n of internal stems. Note that open- and closed-nets-n can be simply related by the addition/deletion of one single-stranded section. Although this classification is quite general, we have limited our numerical studies to all structural topologies that can be decomposed into nets enclosing up to 2 internal helices, which includes most known RNA structures.
Figure 2.

Large-scale description: “Crosslinked gel.” At large scales, we model RNA secondary structures by replacing each constitutive net with a single vertex. These vertices are connected by single-stranded and double-stranded regions. The large-scale conformational entropy is evaluated assuming that the vertices are connected by Gaussian “springs” whose mean squared elongation in isolation equals the relaxed mean squared distance between the connected nets in question. The conformational entropy of such a “Gaussian crosslinked gel” is then calculated numerically by n − 1 algebraic integrations, where n is the number of vertices and hence nets on the secondary structure. (In some cases, two vertices are connected by several stems, in which case we treat them as springs in parallel and lump accordingly.) To better agree with known structures as described in the text, we crudely incorporate excluded volume effects at this large scale by redefining the equilibrium elongation of the “springs” with an excluded volume exponent of 0.65 (vs. 0.5 for the ideal chain).
As an example of short-scale conformational entropy, we present here the analytical result for the canonical pseudoknot (i.e., open-net-2a in Fig. 1; see also Fig. 4b) originally proposed by Pleij et al. (16). Its conformational entropy S depends on the total length of three single-stranded sections (s1, s2, and s3) and on the length of two nonnested helices (l1 and l2) (see Fig. 4b). Modeling the single-stranded sections by Gaussian chains, we find:
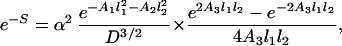 |
1 |
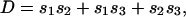 |
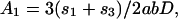 |
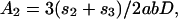 |
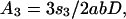 |
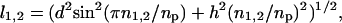 |
where n1,2 are the number of bases in helices 1 and 2, d = 4a is the helix diameter, np = 11 is the number of base pairs per complete turn, and h = 5a is the stem length for one turn (17). α = 0.0068, which is the “confinement” cost each time a loop is formed, has been tuned to best fit the tabulated thermodynamic parameters for short loops (12).
Figure 4.

(a) The secondary structure of the HDV genomic ribozyme (7). (b) The labeling of stems (l1, l2) and single-stranded sections (s1, s2, s3) for the pseudoknot whose entropic free energy is given in Eq. 1; it corresponds to the main pseudoknotted scaffold of the HDV ribozyme (a). (c) The secondary structure of the ribozyme (black) plus attenuator (blue) as it presumably occurs in the HDV genome, except during replication. It corresponds to the absolute free energy minimum obtained by Zuker (ref. 13; http://mfold2.wustl.edu/∼mfold/rna/form1.cgi). Figs. 4, 5, and 7 are drawn by using RnaViz (15).
Note, we employ only the conventional helix free energies (12), and the measured dimensions of paired and single-stranded regions; there are no additional parameters for the pseudoknots, in contrast to earlier phenomenological approaches (3, 18). This physical modeling of pseudoknots is also expected to be more widely applicable than previously proposed estimates (19, 20), as it explicitly takes into account important physical-structural constraints of the RNA molecule, such as those embodied in Eq. 1.
Kinetic Monte Carlo Scheme for RNA.
Each step of the kinetic Monte Carlo algorithm involves making or breaking a stem, and thus a change in topology. For each topology, it often happens that some helices overlap over a few nucleotides on either strand. In such case, a free energy minimization is done to optimally position the boundaries between all competing stems while always leaving a minimum core of Nc bp in each stem. (Typically Nc = 2 or 3 gave very similar kinetics.)
It is also essential to adjust competing helices in any transition configuration, so as to obtain realistic stem formation and dissociation rates. Hence, the barrier ΔG+ for nucleation of a new stem consists of an entropic term involved in bringing two complementary single strands together at a point, plus possibly a free energy term representing the cost of freeing up a minimal stretch of Nc nt when either strand is already engaged in another stem. For each placement of this Nc-long core in the current structure, there can be up to four existing stems in direct overlap with it (Fig. 3a). Considering all possible ways of placing the core (dashed box in Fig. 3.) on the two strands (see parallel dashed lines), we then find the least energetically costly way of shrinking these stems (always keeping the minimum core of Nc bp in any stem) so as to expose the nucleation core. Note that if one strand of the nucleation core falls entirely within a previously formed helix, the latter can be shrunk from either end, which leads to different topologies in the final state, Fig. 3b. The rate is therefore labeled by the topologies of the two states it connects.
Figure 3.

(a) Nucleation core exposed by shrinking up to four overlapping stems. The two horizontal dashed lines highlight the complementary strands of the stem whose formation rates are being evaluated. (b) Each position of the nucleation core may lead to several transition rates corresponding to different topologies.
We complete the calculation of the nucleation rates k+ = k° × exp(−ΔG+/kT) using Gaussian chain statistics to determine the one remaining free constant k° ≃ 108 s−1 from experiments on an isolated stem-loop (5, 6). (In this case, there is no partial unpairing of preexisting stems, so that the Arrhenius rate for each placement of the nucleation core simply reads k°α/s3/2, where s is the length of the corresponding loop.)
Note that, for computation efficiency, the formation rates of new stems involve only local adjustments in helices that directly overlap with the core. For transition rates that remove a stem, however, a global optimization over all of the other competing stems that remain is first done to find the corresponding ground state; the breaking rates k− are then calculated from the reformation rates of the initial stems, k+, as above, modified by the Boltzmann weight between the two ground states, so as to achieve detailed balance. It is computationally tractable to globally optimize the free energy while evaluating breaking rates, because the total number of such transitions is only a small fraction of the number of all possible ways of nucleating new stems. (For example, there are 90 possible helices for the ribozyme in Fig. 4a and over 400 when the “attenuator” sequence is included, Fig. 4c.)
The outline of the final algorithm runs then as follows: For each new structure visited along the folding path, we (i) optimize the boundaries between the competing helices, (ii) recalculate all transition rates (to add or remove single stems), and (iii) select the next transition at random with a probability proportional to its rate. This procedure is then iterated until the time-averaged distribution of the sampled configurations appears stationary, and folding statistics are accumulated by simulating many molecules (typically 30 to several hundred).
It happens frequently during the relaxation that the configuration oscillates repeatedly between a few states, and only much more slowly transits to a new configuration. These local kinetic traps severely impact the efficiency of the code (19). Although these traps can be quite complex, we have found it sufficient to consider only two-state traps (which typically differ by one short stem) for which we can analytically add the rates for all transitions from either trap state to any third state. Thus, we can define a composite move that exits the trap and preserves detailed balance. Since this is exact (no matter how trapped the last two configurations visited are), we in fact use these composite moves continuously.
As the prediction of pseudoknotted structures is not yet settled (3, 20), we have first tested our code on a number of short molecules with proven structures. We obtain good agreement for most examples from refs. 20 and 21. These results are summarized here in the form of the free energy gap predicted between the accepted pseudoknotted structure and the first “excited state,” which differs by at least one helix. For the following RNAs we find: potato leaf roll virus (−2 kT), simian retrovirus 1 (−11 kT), murine leukemia virus (−3 kT), T4 gene 32 (−0.8 kT), mouse mammary tumor virus (−8 kT), and for turnip yellows mosaic virus the accepted pseudoknotted structure is only marginally higher (+0.4 kT) than the ground state we find. However, for tobacco mosaic virus (TMV) the pseudoknotted phylogenetic structure appears to be about 7 kT above the alternative nested ground state we find, and this difference persists for plausible variations in the physical and thermodynamic parameters, such as the helix stacking interactions. Experimental results that probe TMV refolding under the addition of Mg2+ ions (22) suggest that this discrepancy may be because of specific Mg2+ interactions beyond the thermodynamic parameters we used (12).
The current kinetic algorithm needs a few hours of computation on a standard PC to simulate the folding of 200- to 300-nt-long RNA molecules for several minutes. HDV is a single-stranded RNA virus, which contains an 87-nt catalytic piece (Fig. 4a) that refolds into an inactive conformation when attached to a 128-nt attenuator sequence that is contiguous to it in the genome (Fig. 4c) (7, 10). In the remainder of this paper, we present a detailed numerical study of the folding kinetics of this 215-nt-long sequence.
Results: Folding Paths and Regulation of the HDV Ribozyme
The HDV ribozyme and its attenuator sequence are appealing test cases for our algorithm because extensive kinetic data exist for the native sequence as well as many mutants (7, 8); the functional self-cleaving ribozyme is pseudoknotted (Fig. 4a) and active in vitro; and the alternative nonenzymatic conformation, once paired with the attenuator (Fig. 4c), makes the folding kinetics potentially relevant to the efficacy of viral replication in vivo (7, 10).
When we simulated the 87-nt HDV ribozyme, the secondary structure in Fig. 4a represents the lowest free energy state we ever observed, and thus plausibly the globally stable state for our parameters.‖ When quenched from a random coil configuration, about 1/3 of the molecules folded within a fraction of a second to the conformation in Fig. 4a, whereas the remainder were trapped in nonnative conformations for up to a minute (escape time consistent with a single exponential decay statistics). By contrast, when folding occurred simultaneously with regular chain synthesis at 50 nt/s (i.e., the estimated rate for metazoan RNA polymerase II) the fraction of misfolded molecules decreased to less than 10%. Interestingly, at a synthesis rate of 1000 nt/s (the maximum rate for a T7 RNA polymerase used in most in vitro experiments) the misfolded fraction increased to ≈30%. Thus folding is more efficient when coupled to slow synthesis, a fact noted for other systems (11, 24), which suggests that the sequence codes for an efficient folding path, as well as a final structure.
When folding occurs during synthesis, we have found a characteristic bifurcation that occurs within the first ≈40 nt and predicts with high probability whether the molecule in question will be diverted into a kinetic trap when fully synthesized (Fig. 5; see legend for details). Molecules that present both P1 and P5 stems at this stage reach the native structure by the end of synthesis (or shortly after).
Figure 5.

The two competing sets of stems that define the rapid and trapped folding pathways during synthesis. The native stem P1 will tend to nucleate once the first 32 nt are made, provided the current configuration (not drawn) consists of P0 and P5. However when the available sequence is between 26 nt and 31 nt long—i.e., before P1 can nucleate—the alternative stem P6 can form on the structure in conjunction with P0′ (not drawn), and may then become stabilized by another nonnative strong stem, P7. In this event, an inactive misfolded ribozyme is eventually formed at the end of synthesis. It typically consists of stems P6, P4, and P2 plus a sampling of other stems and nucleates P1 only much later if the attenuator is not present. (The same three stems also delineate the trapped species following a quench.) P8 acts as a “folding guide” for both pathways and is easily removed as soon as P4 nucleates and displaces it (see last structures drawn).
Interestingly, at a number of other steps in the sequential pathway, strong stems form that are not part of the final structure but overlap significantly other strong downstream helices. Successive exchanges of stability then occur during synthesis, and the native stems eventually form without any large barriers appearing on the path. For example, we have found that stems P5 and P8 act as transient “folding guides” for the final structure. Indeed, the early formed P5 is first replaced by the combination of P8 and P3 when the sequence length is around 60 nt (Fig. 5). But P5 then reappears (not drawn) once P4 has nucleated and in turn displaces P8 by a simple “zipping–unzipping mechanism” (see Fig. 5). The final native stem to form is P2, which eventually displaces P5 in favor of P3. We note that a stable P5 stem has indeed been recently observed in truncated HDV ribozyme molecules representing several stages along the HDV folding path (see figure 3 in ref. 8), but the role of P8 remains to be confirmed experimentally. Note that this sequential folding process, circumventing large kinetic barriers, is also expected to be fairly insensitive to a large range of synthesis rates, in agreement with our observation (see above). This should ensure, in particular, proper folding with nonconstant—e.g., sequence-dependent—replication rates.
Further evidence that the native sequence is optimized for a synthesis-driven folding comes from the complementary mutations we have tried on P2 and P3, most of which perturb the early optimized folding path, hence increasing the fraction of transiently trapped ribozyme (e.g., C13⋅G82 → A13⋅U82 (≈30%) or G17⋅C30 → C17⋅G30 (≈30%)). On the contrary, complementary mutations in the top 3 bp of P1 have little negative effect or even increase the fast folding fraction to nearly 100% (e.g., G6⋅C32 → A6⋅U32) because they do not disrupt the early folding path (i.e., P0, P5, P1) while weakening P6 with an internal mismatch.
As expected, however, the effect of a specific mutation is difficult to predict a priori. An additional base pair added to the top of P1 could act by means of the stem length and the conformational entropy (25), but instead operates through the alternative folding paths in Fig. 5. Indeed, a U⋅A base pair has a minor effect on the folding kinetics, whereas adding a G⋅C base pair at the same location increases the length of P6 by 2 bp and facilitates its nucleation (≈30%) via a new “kissing hairpin-loops” transition (26) from the stable intermediate structure consisting of stems P0 and P5. It also lowers the free energy of the trapped misfolded structure (−83.6 kT) below that of the catalytic one (−80.6 kT), which may explain the poor enzymatic activity seen in experiments (25).
Investigating for possible influence of the 20-nt-long sequence upstream of the cleavage site (Fig. 4a), we observed that the wild-type 5′ tail (5′-UUCCAUCCUUUCUUACCUGAU-3′) essentially behaves as an inert “spacer” preventing cross-hybridization between the upstream sequence and the ribozyme. However, a strongly mutated 5′ tail can potentially prevent the ribozyme from folding correctly (e.g., 5′-UUCCAUCuUgUCUagCugGAU-3′ strongly interferes with P1).
The “folding chart” in Fig. 6 allows one to visualize in a single image the more complex folding pathways of the 215-nt ribozyme plus attenuator sequence. For either pathway in Fig. 5, we find that essential single-stranded regions of the ribozyme's catalytic core hybridize with part of the attenuator, as soon as it is being synthesized (Figs. 6 and 7). Thus for the virus, there is a short window of times during which self-cleavage must occur in the context of the “double-rolling-circle” mechanism by which HDV replicates (7). Failure to rapidly cleave with 100% efficiency should lead to the formation of a few mainly dimeric copies, which indeed have been detected in vivo (10). The globular structure in Fig. 7 then evolves on a time of several minutes to the linear structure of minimum free energy shown in Fig. 4c.
Figure 6.

Chart of helices present in the ribozyme plus attenuator sequence as it folds during and after synthesis for a molecule that folds via the catalytic folding path in Fig. 5 (red), and a molecule following the main noncatalytic path in Fig. 5 (green). At each time, a point is marked for the 3′ end of all helices present on the structure (with red and green slightly offset), thus isolated points are transient helices and continuous lines, stable ones. Synthesis is complete at 4 s, and the labeled stems follow the numbering of Fig. 5. The last intermediate structures drawn in Fig. 5, when the top of P4 has just nucleated and starts displacing P8, correspond to the time indicated by the arrow.
Figure 7.

The secondary structure corresponding to the red curve at 10 s in Fig. 6 (i.e., catalytic path). The native ribozyme stems are still present, but various single-stranded regions (red) have already paired with the attenuator sequence (blue), presumably preventing further catalytic activity.
Our complementary study of the HDV antigenomic ribozyme (7) confirms recent experiments proposing an elongated P2 stem (27). The lowest free energy configurations we find for this RNA contain, however, a 1-bp mismatch and a 2-bp elongation on top of P4. Although this extension overlaps the catalytic core of the ribozyme, it is weak enough (ca. −5 kT) to be removed by thermal fluctuations. However, if the mismatch is corrected (e.g., G42⋅G75 → C42⋅G75), the strain induced on the pseudoknot by the longer and stronger P4 breaks P3, the breaking presumably coinciding with the loss of enzymatic activity observed in vitro (27).
Discussion
Although our code does relax to the accepted ground state for a number of RNA molecules and provides plausible kinetic explanations for a number of experiments, it is only a heuristic tool. Stem free energies change by ∼15% between different releases of the parameter sets (http://mfold2.wustl.edu/∼mfold/rna/form1.cgi); we use only approximate parameters for stacked helices; there is no excluded volume effect between nearby single strands and stems; and all of the chemical specificity of the three-dimensional structure is lost. Certain results, such as the dynamic bifurcation and the transient guiding stems we have found, are fairly insensitive to such details, as they remain unchanged for plausible changes in thermodynamic and structural parameters. But numbers such as individual kinetic rates and the overall folding time can easily be off by a factor of 10. Still, simulations promise to be an important adjunct to the many detailed studies of RNA folding now underway (8, 9), as they can reach comparable times and rapidly generate hypothesis as to how sequence changes affect structure and kinetics, in particular transient guiding stems on the folding path.
For our simulations of the HDV ribozyme, we find that a fraction of the molecules fold rapidly (i.e., by the end of synthesis when the two processes occur simultaneously), whereas the remainder are trapped. The distribution of folding times for the trapped fraction is consistent with a single-exponential decay statistics (i.e., standard deviation equals mean), whereas the average folding time decreases with increasing temperature. Both results are consistent with a single thermally activated rate-limiting step required to exit the trap, in contrast to the general downward trend with no large barriers we have found for the main sequential folding path. These are all issues that have arisen in protein folding (28, 29), and our findings for the HDV ribozyme are similar to the consensus for proteins.
Indeed, vastly more theory and simulation studies have been devoted to the folding of proteins than to RNAs because of their central place in biochemistry and medicine. Yet, the RNA problem is technically simpler because the self-paired stems are better defined energetically than the corresponding α-helices and β-sheets of proteins. Independent experimental studies (1, 5, 9) also strongly support a processive folding kinetics for RNAs, with relatively well-defined transition states and rates between the intermediate folded structures. Hence, in comparison with the much more cooperative folding dynamics of proteins, these simpler folding kinetics of RNAs can be easily used in kinetic algorithms such as ours, which ultimately provide more realistic predictions on interesting time scales.
Such code can also be readily adapted to include a force applied to the two ends of the molecule, hence modeling the succession of structures realized in a mechanical unfolding experiment. Measurements of force vs. distance under these conditions may provide interesting insights on RNA structures, but their interpretation will require theory. Finally, similar kinetic simulations may also provide a quantitative understanding of hybridization kinetics, which is an issue very relevant to the performance of DNA microarrays (30) and the efficacy of artificial antisense RNAs for therapeutics (31).
Acknowledgments
We thank M. D. Been, M. Newman, N. Socci, E. Westhof, and G. S. Wickham for discussions. H.I. was supported in succession by Cornell University, a Lavoisier Fellowship from the French Foreign Ministry, a National Institutes of Health grant, and the Klaus Tschira Foundation.
Abbreviation
- HDV
hepatitis delta virus
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.110533697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.110533697
References
- 1.Brion P, Westhof E. Annu Rev Biophys Biomol Struct. 1997;26:113–137. doi: 10.1146/annurev.biophys.26.1.113. [DOI] [PubMed] [Google Scholar]
- 2.Nussinov R, Jacobson A B. Proc Natl Acad Sci USA. 1980;77:7826–7830. doi: 10.1073/pnas.77.11.6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rivas E, Eddy S R. J Mol Biol. 1999;285:2053–2068. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- 4.Mironov A A, Dyakonova L P, Kister A E. J Biomol Struct Dyn. 1985;2:953–962. doi: 10.1080/07391102.1985.10507611. [DOI] [PubMed] [Google Scholar]
- 5.Pörschke D. Biophys Chem. 1974;1:381–386. doi: 10.1016/0301-4622(74)85008-8. [DOI] [PubMed] [Google Scholar]
- 6.Bonnet G, Krichevsky O, Libchaber A. Proc Natl Acad Sci USA. 1998;95:8602–8606. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Been M D, Wickham G S. Eur J Biochem. 1997;247:741–753. doi: 10.1111/j.1432-1033.1997.00741.x. [DOI] [PubMed] [Google Scholar]
- 8.Matysiak M, Wrzesinski J, Ciesiolka J. J Mol Biol. 1999;291:283–294. doi: 10.1006/jmbi.1999.2955. [DOI] [PubMed] [Google Scholar]
- 9.Treiber D K, Rook M S, Zarrinkar P P, Williamson J R. Science. 1998;279:1943–1946. doi: 10.1126/science.279.5358.1943. [DOI] [PubMed] [Google Scholar]
- 10.Lai M M C. Annu Rev Biochem. 1995;64:259–286. doi: 10.1146/annurev.bi.64.070195.001355. [DOI] [PubMed] [Google Scholar]
- 11.Morgan S R, Higgs P G. J Chem Phys. 1996;105:7152–7157. [Google Scholar]
- 12.Serra M J, Turner D H, Freier S M. Methods Enzymol. 1995;259:243–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 13.Zuker M. Methods Enzymol. 1989;180:262–288. doi: 10.1016/0076-6879(89)80106-5. [DOI] [PubMed] [Google Scholar]
- 14.Smith S B, Cui Y, Bustamante C. Science. 1996;271:795–799. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 15.De Rijk P, De Wachter R. Nucleic Acids Res. 1997;25:4679–4684. doi: 10.1093/nar/25.22.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pleij C W A, Rietveld K, Bosch L. Nucleic Acids Res. 1985;13:1717–1731. doi: 10.1093/nar/13.5.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bloomfield V A, Crothers D M, Tinoco I., Jr . Physical Chemistry of Nucleic Acids. New York: Harper & Row; 1974. [Google Scholar]
- 18.Abrahams J P, van den Berg M, van Batenburg E, Pleij C W A. Nucleic Acids Res. 1990;18:3035–3044. doi: 10.1093/nar/18.10.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mironov A A, Lebedev V F. BioSystems. 1993;30:49–56. doi: 10.1016/0303-2647(93)90061-g. [DOI] [PubMed] [Google Scholar]
- 20.Gultyaev A P, van Batenburg E, Pleij C W A. RNA. 1999;5:609–617. doi: 10.1017/s135583829998189x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang H, Hines J H, Tinoco I., Jr J Mol Biol. 1996;259:135–147. doi: 10.1006/jmbi.1996.0308. [DOI] [PubMed] [Google Scholar]
- 22.van Belkum A, Abrahams J P, Pleij C W A, Bosch L. Nucleic Acids Res. 1985;13:7673–7386. doi: 10.1093/nar/13.21.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferre-D'Amare A R, Zhou K, Doudna J A. Nature (London) 1998;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- 24.Gultyaev A P, van Batenburg E, Pleij C W A. J Mol Biol. 1995;250:37–51. doi: 10.1006/jmbi.1995.0356. [DOI] [PubMed] [Google Scholar]
- 25.Tanner N K, Schaff S, Thill G, Petit-Koskas E, Crain-Denoyelle A-M, Westhof E. Curr Biol. 1994;4:488–498. doi: 10.1016/s0960-9822(00)00109-3. [DOI] [PubMed] [Google Scholar]
- 26.Tinoco I., Jr Nucleic Acids Symp Ser. 1997;36:49–51. [PubMed] [Google Scholar]
- 27.Perrotta A T, Nikiforova O, Been M D. Nucleic Acids Res. 1999;27:795–802. doi: 10.1093/nar/27.3.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Onuchic J N, Luthey-Schulten Z, Wolynes P G. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 29.Thirumalai D, Woodson S A. Acc Chem Res. 1996;29:433–439. [Google Scholar]
- 30.Mir K U, Southern E M. Nat Biotechnol. 1999;8:788–792. doi: 10.1038/11732. [DOI] [PubMed] [Google Scholar]
- 31.Patzel V, Sczakiel G. Nat Biotechnol. 1998;16:64–68. doi: 10.1038/nbt0198-64. [DOI] [PubMed] [Google Scholar]