

Abstract
Background
The 13q-deletion syndrome causes human congenital birth defects due to the loss of regions of one long arm of human chromosome 13. A distal critical region for severe genitourinary and anorectal birth defects in the region of 13q32.2-34 has been suggested; we sought to narrow this critical region.
Methods
From patients with karyotypes revealing haploinsufficiency for distal chromosome 13q and their parents, peripheral blood was obtained and lymphocytes were immortalized for DNA isolation. Genetic and molecular cytogenetic methods were used to map deletions. Patient and parental samples were genotyped with a panel of 20 microsatellite markers spanning 13q31.3 qter and deletions identified by loss of heterozygosity. Deletions were also mapped using a panel of 35 BAC clones from the same region as probes for fluorescence in-situ hybridization on patient lymphoblastoid metaphase preparations. The data were synthesized and a deletion map defining the critical region was generated.
Results
Eight patients with known deletions around 13q32qter and their parents were analyzed, and categorized into three groups: three patients with anorectal and genitourinary anomalies (hypospadias, penoscrotal transposition), four male patients without anorectal and genitourinary anomalies, and one XY patient with ambiguous genitalia without anorectal anomalies. We mapped the critical region for anorectal and genitourinary anomalies to a ∼9.5-Mb interval of 13q33.3-q34 delineated by markers D13S280-D13S285; this spans ∼8% of the chromosome and contains 20 annotated genes
Conclusion
The critical region of chromosome 13q mediating genitourinary/anorectal anomalies has been mapped, and will be narrowed by additional patients and further mapping. Identification of the gene(s) mediating these syndromic genitourinary defects should further our knowledge of molecular mediators of non-syndromic hypospadias, penoscrotal transposition and anorectal malformations.
Keywords: Chromosome, 13q, Hypospadias, Penoscrotal Transposition, Anorectal malformation, Deletion, Haploinsufficiency
Introduction
The molecular mechanisms and factors that govern embryological morphological events such as penoscrotal positioning, urethral tubularization, cloacal septation, and closure of the perineum and scrotum are not fully understood. In order to identify new molecular mediators of these developmental processes in humans, we investigated the molecular basis of genital and anorectal abnormalities in patients with the chromosome 13q-deletion syndrome. This syndrome 1, or loss of regions of one long arm of human chromosome 13, causes major human congenital birth defects, including severe genitourinary and anorectal malformations (Figures 1 and 2). Bartsch and colleagues 2, 3 described deletions of 13q (q33.2-qter) associated with penoscrotal inversion, hypospadias, reduced anogenital distance, imperforate anus, facial anomalies and developmental delay. They suggested the presence of a critical gene(s) in 13q32.2-q34 mediating these genitourinary and anorectal anomalies1. Additional phenotypes associated with distal 13q deletion in the literature include ambiguous genitalia in males4-6, and absent or bicornuate uterus, imperforate anus with vaginal fistula, or cloaca in females7-9. Thus, haploinsufficiency of one or more distal 13q genes appears to cause these phenotypes.
Figure 1.
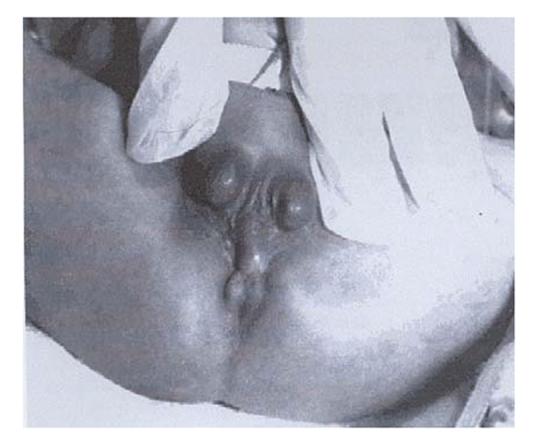
The perineum of a 46, XY, del (13)(q31.1) male with complete penoscrotal transposition, perineal hypospadias and imperforate anus (Walsh et al., 200117).
Figure 2.
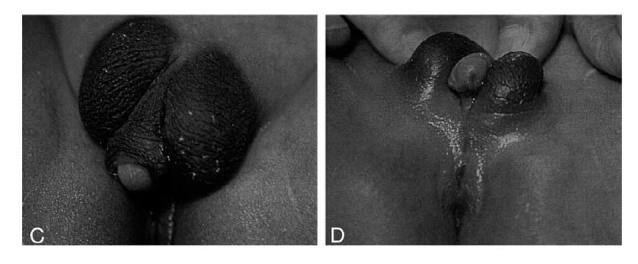
The external genitalia and the perineum of a male with mosaicism of ring chromosome 13 and monosomy 13q del q33.2-qter (Kuhnle et al., 2000 3).
Each of these birth defects, when independently assessed, is incompletely understood at the embryological and molecular level. In fact, despite the frequency of these individual birth defects in the general population, the etiology is unknown in the majority of human cases. Several studies conclude that the incidence of hypospadias is increasing 10 possibly due to environmental exposures to ‘endocrine disruptors’ 11-13. Hypospadias is the second most common human birth defect, with an incidence of 1 in 125 male births 14. Given the increasing incidence and significant morbidity associated with these birth defects, the importance of understanding embryonic and molecular mechanisms of normal and abnormal development has emerged as a key issue.
Although cytogenetic karyotyping is standard testing for children with multiple congenital birth defects, the accuracy and resolution of classic cytogenetic karyotyping is limited to changes >5 Mb. To improve resolution, we mapped distal 13q deletions using molecular markers and examined associated phenotypes in order to refine the critical region(s) for urogenital and anorectal anomalies and identify candidate gene(s) for these birth defects.
Materials and Methods
Patient recruitment and phenotype analysis
Using University of Texas Southwestern Institutional Review Board approved methods, patients were ascertained with karyotypes revealing haploinsufficiency for distal chromosome 13q (distal 13q deletion) via medical record review and search of cytogenetics databases. Patients and their family members were recruited through local sources and international collaborators. Detailed phenotypic data were collected from interviews with parents and physicians as well as from medical records. When available, local subjects were clinically evaluated by the senior author, an experienced pediatric urologist, documenting phenotype.
Genotype mapping
Peripheral blood samples were collected from probands, their available parents, and any other affected family members. From proband blood, Epstein-Barr virus-immortalized lymphoblastoid cell lines were generated by standard methods. Genomic DNA was extracted from all remaining family members’ blood samples. 13q deletions were mapped to high resolution by testing for loss of heterozygosity (LOH) of 20 polymorphic microsatellite markers from the ABI PRISM® Linkage Mapping Set version 2.5 (Applied Biosystems, Foster City, CA, USA), supplemented by custom markers chosen from the genome database (www.gdb.org). One marker was specifically designed to amplify a polymorphic CA dinucleotide repeat within the ephrin-B2 (EFNB2) gene. For samples without parental DNAs or where key markers were not informative, fluorescence in-situ hybridization (FISH) was performed on proband lymphoblastoid metaphase preparations using probes from our panel of 35 BAC clones from the same chromosomal region (BACPAC Resource Center, Children’s Hospital Oakland Research Institute, Oakland, CA, USA; (http://bacpac.chori.org/).
Determination of critical region by genotype-phenotype correlation
The data were synthesized and a deletion map defining the critical region was generated. Critical regions for urogenital and anorectal phenotypes were defined by the minimal overlap among deletions in patients with urogenital and/or anorectal phenotypes. With the possible exception of hypospadias, which is relatively common, 13q-patients with a genitourinary or anorectal anomaly should be deleted for the critical region for that phenotype. However, every patient who is deleted for the critical region will not necessarily have the associated phenotype, because haploinsufficiency of the culprit gene may show incomplete penetrance. Thus, we defined the critical region by comparing the deletions among only those patients with the phenotype in question.
Generation of the candidate gene list from annotated databases
A refined list of genes in the critical region was generated from the most current version of the UCSC Genome Browser. Functional genomic databases (e.g. GeneCards) and the PubMed literature database were searched to evaluate current knowledge about these genes regarding embryonic expression and known or suspected function in developmental signaling pathways.
Results
Phenotypic analysis of 13q-patients
Ten probands (9M:1F determined by sex chromosomal complement) with known deletions around 13q32-qter and 24 family members (parents, siblings, and grandparents) were recruited (Table 1). Of these, eight were included in this analysis, excluding one female and one male whose 13q deletions proved to be proximal to our region of interest. Phenotypic analysis is displayed in Table 2. Of these eight probands, six were newly ascertained for this study; one sample (#7) was obtained from the NIGMS Camden Repository (ccr.coriell.org/nigms), and one previously reported sample (#6) 2 was a kind gift obtained from Dr Oliver Bartsch. These eight probands could be classified into three phenotypic groups: (1) three male patients with developmental delay and without anorectal and genitourinary anomalies (#1-3); (2) four patients with anorectal malformation or anorectal malformation + genitourinary anomalies (hypospadias, penoscrotal transposition) (#4-7); and (3) one XY patient with ambiguous genitalia without anorectal anomalies (#8).
Table 1.
Sex and racial distribution of the 13q-deletion study participants
| ETHNICITY/GENDER | 13q Deletion probands | 13Q Family members | TOTAL |
|---|---|---|---|
| Caucasian (MALE) | 3 | 5 | 8 |
| (FEMALE) | 1 | 8 | 9 |
| African-American (MALE) | |||
| (FEMALE) | |||
| Hispanic (MALE) | 5 | 4 | 9 |
| (FEMALE) | 7 | 7 | |
| Asian (MALE) | |||
| (FEMALE) | |||
| Other (MALE) | |||
| (FEMALE) | |||
| Unknown | 1 | 1 | |
| TOTAL | 10 | 24 | 34 |
Table 2.
Phenotypic characterization of eight XY patients with known 13q32-34 deletions
| Patient no. | Group | Sex | Karyotype | Phenotype |
|---|---|---|---|---|
| 1 | 1 | M | ring 13q | Bilateral VUR, dysplastic upper tracts, meningomyelocoele, microcephaly and developmental delay |
| 2 | 1 | M | 46,XY del 13q33.2 | No genitourinary anomalies, |
| microcephaly | ||||
| 3 | 1 | M | 46,XY del 13q33.2 | Right ureteral duplication, no genital or anal anomalies |
| 4 | 2 | M | 46,XY del 13q33.2 | Anorectal malformation |
| 5 | 2 | M | 46,XY del13q33 | Bilateral cleft lip and palate, anteriorly displaced anus, hypospadias |
| 6 | 2 | M | 45,XY,-13,-22,+der(13:22)(q32.2;p11) | Perineal hypospadias, penoscrotal transposition, bifid scrotum, bilaterally descended testes, low anteriorly displaced imperforate anus |
| 7 | 2 | M | 46,XY,-13,+der(13)t(6;13)(13pter>13q32::6q27>6qter)mat | Bifid scrotum, displaced anus, micropenis |
| 8 | 3 | Intersex, raised female | 46,XY,der(13)t(6;13)(p25;q33) | Ambiguous genitalia (Male pseudohermaphrodite), no anal anomaly |
Deletion mapping of 13q critical region for urogenital and anorectal phenotypes-Candidate Gene List
LOH and FISH results (Fig. 3) were concordant in cases where both analyses were performed. Due to limited quantities of DNA for proband #6, the proximal limit of the deletion in the proband was obtained from the literature. The deletion in this patient is unlikely to be smaller than the deletion in proband #4 or #8, and thus will probably not narrow the critical region. Data synthesis to date has yielded a deletion map defining the 13q critical region mediating genitourinary/anorectal anomalies to an ∼11-Mb interval distal to marker D13S280 (Fig. 4). Five of the eight subjects have either anorectal or urogenital abnormalities, and three have both. The minimal overlapping deletion for both phenotypes (#4-anorectal malformation; #8-ambiguous genitalia/male pseudohermaphrodite) is proximally delimited by marker D13S280 in 13q33.1, making the critical region D13S280-D13S285, an interval of 9.5 Mb. This assumption is based on the deletion noted on patient #3, but a possible lack of penetrance of urogenital/anorectal malformations should be considered in this patient. This would then make the critical region D13S280-qtel, an interval of 11 Mb. It may be possible to narrow the anorectal anomaly critical region further by genotyping additional microsatellite markers or by FISH studies in #4, as D13S158 and D13S274 were uninformative. The breakpoint in #8 is already mapped to an ∼400-kb interval between markers D13S280 and D13S158. As shown in Fig. 4, anorectal malformation is associated with both maternal and paternal 13q deletions, suggesting that this phenotype does not involve an imprinted locus. At present, four out of four deletions associated with urogenital abnormalities are paternal, but the numbers are too small to know whether this phenotype involves a maternally imprinted gene. From this deletion map, 20 annotated genes are identified in the interval D13S280-D13S285, based on the UC Santa Cruz Genome Browser annotation (May 2004 Freeze) (Table 3).
Figure 3.

FISH on lymphoblastoid metaphase preps. Green probe is BAC AL138689.21, containing the ephrin-B2 gene (EFNB2). Red probe is retinoblastoma gene (RB) control. (A) Proband #7-EFNB2 is deleted. (B) Proband #3-EFNB2 is not deleted.
Figure 4.
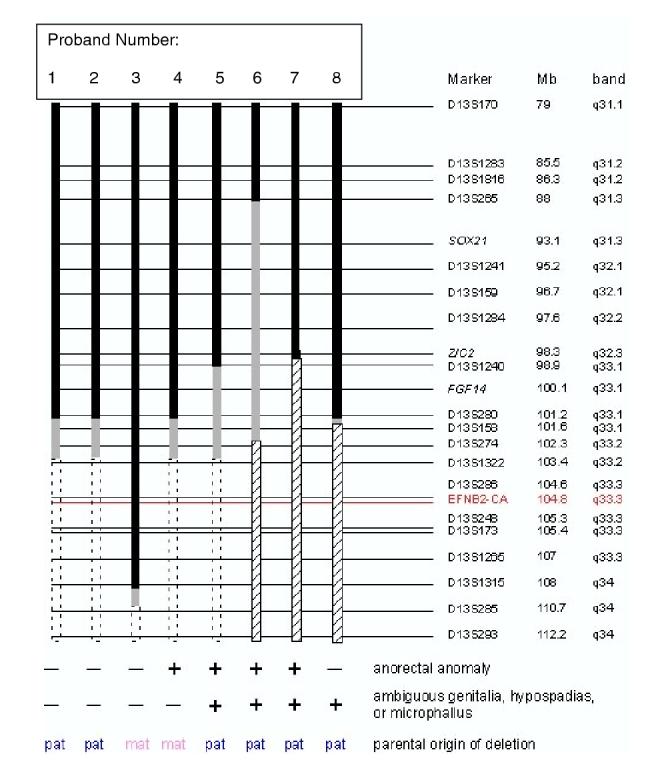
Deletion mapping of 13q-patients. Solid black bars indicate 13q sequences that are not deleted; dashed open boxes denote deletions; gray bars indicate regions of uncertainty; hatched bars indicate deletions due to unbalanced translocations. Loci names and physical and cytogenetic map locations of microsatellite markers and selected FISH probes (italics) according to the UC Santa Cruz Genome Browser (genome.ucsc.edu, July 2003 assembly) are indicated. EFNB2-CA marker is indicated in red. Presence (+) or absence (-) of phenotypes and parental origin (pat = paternal; mat = maternal) of deletions are listed.
Table 3.
The 20 annotated genes in the interval D13S280-13qter, based on the UC Santa Cruz Genome Browser annotation (May 2004 Freeze)
| Candidate gene name | Description | Comment |
|---|---|---|
| SLC10A2 | Ileal sodium/bile acid cotransporter | Mutations cause 1° bile acid malabsorption |
| DAOA | D-amino acid oxidase activator | |
| EFNB2 | pEhrin B2 | Cell-cell signaling; neural and vascular development; hypospadias and anorectal malformation in mice46 |
| FLJ10154 | Hypothetical protein | Unknown function |
| LIG4 | DNA ligase 4 | Functions in immunoglobulin gene rearrangement |
| C13orf6 | Novel protein | Esterase motif |
| TNFSF13B | TNF ligand superfamily | B cell cytokine |
| KIAA0865 | Novel protein | Expressed in brain |
| IRS2 | Insulin receptor substrate 2 | Implicated in type II diabetes |
| COL4A1 | alpha 1 type IV collagen preproprotein | Structural component of kidney glomerulus basement membrane |
| COL4A2 | alpha 2 type IV collagen preproprotein | Structural component of kidney glomerulus basement membrane |
| RAB20 | RAS oncogene family member | Plays a role in apical endocytosis/recycling |
| FLJ10769 | Hypothetical protein | Unknown function |
| FLJ12118 | Hypothetical protein | cysteinyl-tRNA synthase motif |
| ING1 | Inhibitor of growth family member 1 | Tumor suppressor; interacts with p53 |
| LOC283487 | Hypothetical protein | Unknown function |
| ANKRD10 | Ankryn repeat domain 10 | |
| ARHGEF7 | Rho guanine nucleotide exchange factor 7 isoform | Induces cellular membrane ruffling |
| MGC35169 | Hypothetical protein | Actin crosslinking domain |
| SOX1 | SRY-box 1 gene | Transcription factor; mouse mutation causes epilepsy and abnormal brain development |
Discussion
Human chromosomal alterations can range from those detectable at the karyotypic level to point mutations. This translates to a broad range of phenotypic variability, especially when one considers the vast number of mechanisms that regulate gene expression. While this phenotypic variability complicates the analysis, cases with chromosomal or gene alterations are ‘experiments by nature’ that have led to elucidation of the genetic basis of many congenital birth defects.
Originally designated as one of the D-group autosomes, human chromosome 13 is depicted in Fig. 4 with its Giemsa banding nomenclature. In 1969, Allderdice and colleagues 1 described the ‘Chromosome 13q-Syndrome’. A list of the many phenotypes described in this syndrome is presented in Table 4; the exact phenotypes expressed are related to the chromosomal segment(s) deleted. Investigations of monosomy 13 patients with retinoblastoma or holoprosencephaly resulted in the positional cloning of the retinoblastoma gene (Rb) at 13q14 15 and the ZIC2 gene 16 at 13q32, respectively. From a review of 20 patients with 13q deletions, Brown et al. 7 concluded that proximal deletions (q13-q31) are associated with mental retardation without major deformities except for retinoblastoma, while deletions involving band 13q32 are usually associated with numerous major malformations. They reported that deletions from q33-qter typically manifest severe mental retardation with more minor abnormalities; however, they did not include genitourinary/anorectal anomalies as a major malformation. Unfortunately, one problem with karyotype/phenotype correlations is interpreting banding patterns of deleted chromosomes. As reported by Brown et al., “it is particularly difficult to decide, in the case of distal 13q deletions involving loss of the Giemsa-dark band q33, whether a remaining Giemsa-light band represents part of q32 or q34 or both” 7. These authors recommended molecular markers to define accurately the exact deletions in the distal 13q region.
Table 4.
Common phenotypes observed in the human chromosome 13q-deletion syndrome
| Phenotype | Frequency |
|---|---|
| Psychomotor retardation | 94% |
| Hypertelorism, microcephaly | 94% |
| Holoprosencephaly - ZIC2 gene at 13q32 | |
| Agenesis of the corpus callosum | |
| Prominent nasofrontal bones | 66% |
| Ear abnormalities | 79% |
| Microphthalmia/coloboma of the iris | 25% |
| Retinoblastoma - RB1 gene at 13q14.1-q14.2 | 18% |
| High arched or cleft palate | |
| Congenital heart disease (atrial septal defect, ventricular septal defect, tetralogy of Fallot, patent ductus arteriosus, aortic coarctation) | 55% |
| Factor VII and X clotting deficiency | |
| Hypoplasia or aplasia of the thumbs/toes, syndactyly, brachydactyly | 27% |
| Duodenal atresia, intestinal malrotation, Hirschsprung’s disease | |
| Renal hypoplasia, hydronephrosis | |
| Biseptate or absent uterus, cloaca | |
| Imperforate or anteriorly displaced anus* | 16% |
| Genital ambiguity* | |
| Hypospadias, penoscrotal transposition, bifid scrotum* | 38% |
| Neural tube defects |
Genitourinary/anorectal phenotypes. When known, causal genes, their chromosomal location, and the frequency of the anomaly are noted 48.48
Two reports by Bartsch and colleagues 2, 3 of the clinical syndrome of penoscrotal inversion, hypospadias, reduced anogenital distance, imperforate anus, facial anomalies and developmental delay in distal chromosome 13q deletions (Online Mendelian Inheritance in Man # 602553) led these authors to suggest a critical region 13q33.2-qter mediating these defects. As seen in Figs 1 and 2, the external genital phenotype of the male child with a distal deletion in chromosome 13q is variable in severity3, 17. Some case reports have indicated ambiguous genitalia in XY cases of 13q deletion syndrome4-6;however, it is not clear by what criteria this term was used, thus adding to some phenotypic confusion in the literature. Nevertheless, the male phenotype is severe and some die in utero or soon after birth18. In females, absent or bicornuate uterus, imperforate anus with vaginal fistula, and cloaca have been reported7-9.
In this study, we focused on the male urogenital/anorectal manifestations of the 13q deletion syndrome. Hypospadias as an isolated defect is clearly a common human disorder with mild to severe phenotypic variability. Fortunately, proximal hypospadias accounts for about 15% of cases. In addition, proximal hypospadias cases can be associated with additional malformations, including penoscrotal transposition and anorectal malformations. The literature reports that up to 2.2%-10.5% of patients with anorectal malformations also have hypospadias 19-21. Penoscrotal transposition, wherein the scrotum is partially or completely above the penis (see Figs 1 and 2), is a severe anomaly most commonly associated with severe hypospadias. This birth defect is strongly associated with deletions of chromosome 13q. Forty-six PubMed citations discuss penoscrotal transposition and, of these, six of 11 with chromosomal anomalies had 13q distal deletions 2, 17, 22-25. Penoscrotal transposition is associated with imperforate anus in about a third of cases 26. Anorectal malformations also demonstrate great phenotypic variability, affecting 1 in 1500 to 1 in 5000 live births, with an even gender distribution 27-30. Although a male patient with this constellation of birth defects is a rare case, molecular based investigations may lead to the elucidation of key developmental molecular mechanisms not yet understood. This suggests that an alteration of human chromosome 13 could account for a significant proportion of patients with anorectal malformations and hypospadias/penoscrotal transposition.
Animal models of hypospadias and anorectal malformations have been genetically engineered and can be created by drug exposure. Hypospadias has been noted in mouse models mutant for Shh31, 32, Bmp’s and Fgf’s33, Hoxa1334-36, Hoxd1337, and in retinoic acid-treated animals38, 39. Several genes have been implicated in anorectal malformations in mutant mouse models, namely Sonic hedgehog (Shh) 40-43, Gli 44, 45 and ephrin-B2(Efnb2) 46. An ephrin-B2 homozygous null mutation is embryonic lethal due to defects in angiogenesis, and no heterozygous phenotype was reported 47; however, our group generated a partial loss of function mutation that interferes with ephrin signaling, ephrin-B2 lacZ/+, which revealed hypospadias in heterozygous males at 40% penetrance 46. The striking phenotype of male imperforate anus (see Fig. 6) and female persistent cloaca was seen in the homozygous ephrin-B2 lacZ/lacZ mice at 100% penetrance 46. The observations of this mutant mouse phenotype and the fact that the human ephrin-B2 gene (EFNB2) is located at 13q33.3 has prompted us to investigate the role of EFNB2 in human hypospadias and in the 13q deletion syndrome.
Figure 6.

Left panel reveals a sagittal H&E-stained section of an embryonic day 18 wild-type male mouse with normal genitourinary and anorectal development. Right panel contrasts the embryonic day 18 ephrin-B2 lacZ/lacZ male littermate with malformed urethra, high imperforate anus and abnormal colonic connection to the base of the bladder (rectovesical fistula). From Dravis et al. 2004 46.
In our study of eight genetically male probands with distal 13q deletion syndrome, five patients manifest urogenital/anorectal malformations and three do not. Our molecular mapping of their 13q deletions reveals that the minimal critical region for anorectal and urogenital anomalies is delimited by microsatellite markers D13S280-D13S285, approximately 9.5-11 Mb, when considering possible lack of penetrance in patient 3. This corresponds to 13q33.1-q34/tel, 3.5-5 Mb smaller than the critical region previously defined cytogenetically2. The critical region contains 20 annotated genes, including EFNB2. Based on our literature review, this gene appears to be the top candidate causal gene. Both urogenital and anorectal anomalies are seen with either maternal or paternal deletions, implying that imprinting does not play a role in these phenotypes. Given these urogenital and anorectal anomalies show incomplete penetrance in 13q-patients, a larger sample of male patients will need to be studied to further refine the critical region and confirm or refute the role of EFNB2 in the urogenital/anorectal phenotype of the 13q-deletion syndrome. The recent introduction of molecular karyotyping via microarray comparative genomic hybridization may further validate this work with much greater resolution.
Conclusions
The critical region of chromosome 13q mediating genitourinary/anorectal anomalies has been further refined to a an ∼9.5 Mb interval containing 20 annotated genes. Haploinsufficiency of 13q shows incomplete penetrance for anorectal and genitourinary anomalies. Recruitment and mapping of additional 13q-cases will further narrow the critical region. Identification of the gene(s) mediating these syndromic genitourinary defects will likely further our knowledge of molecular mediators of non-syndromic hypospadias, penoscrotal transposition and anorectal malformations.
Figure 5.

Idiogram of chromosome 13. Red bar indicates critical region.
Acknowledgements
We thank Dr Oliver Bartsch and Dr Batanian for providing each one sample and their respective families. We also thank the patients and families who participated at Children’s Medical Center of Dallas as well as Jocelyn Allgood, R.N. and Robert Barber, R.N. for collecting samples. Supported in part by the NIH R01 DK59164 (PI: Baker, Linda A.) and NIH R01 DK59164-S1 (Baker/Garcia), the Seay Pediatric Urology Endowment (Baker), and the Bob Smith Children’s Medical Center Endowment (Baker).
Footnotes
Funded in part by: NIH R01 DK59164 (PI: Baker, Linda A.) and NIH R01 DK59164-S1 (Baker/Garcia), UTSW Seay Pediatric Urology Endowment, Bob Smith Children’s Medical Center at Dallas Endowment.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allderdice PW, Davis JG, Miller OJ, Klinger HP, Warburton D, Miller DA, et al. The 13q-deletion syndrome. Nature. 1969:499. [PMC free article] [PubMed] [Google Scholar]
- 2.Bartsch O, Kuhnle U, Wu LL, Schwinger E, Hinkel GK. Evidence for a critical region for penoscrotal inversion, hypospadias, and imperforate anus within chromosomal region 13q32.2q34. Am J Med Genet. 1996;65:218. doi: 10.1002/(SICI)1096-8628(19961028)65:3<218::AID-AJMG9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 3.Kuhnle U, Bartsch O, Werner W, Schuster T. Penoscrotal inversion, hypospadias, imperforate anus, facial anomalies, and developmental delay: definition of a new clinical syndrome. Pediatr Surg Int. 2000;16:396. doi: 10.1007/s003830000379. [DOI] [PubMed] [Google Scholar]
- 4.Luo J, Balkin N, Stewart JF, Sarwark JF, Charrow J, Nye JS. Neural tube defects and the 13q deletion syndrome: evidence for a critical region in 13q33-34. Am J Med Genet. 2000;91:227. doi: 10.1002/(sici)1096-8628(20000320)91:3<227::aid-ajmg14>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 5.Brown S, Russo J, Chitayat D, Warburton D. The 13q syndrome: the molecular definition of a critical deletion region in band 13q32. Am. J. Hum. Genet. 1995;57:859. [PMC free article] [PubMed] [Google Scholar]
- 6.Gutierrez J, Sepulveda W, Saez R, Carstens E, Sanchez J. Prenatal diagnosis of 13q-syndrome in a fetus with holoprosencephaly and thumb agenesis. Ultrasound Obstet Gynecol. 2001;17:166. doi: 10.1046/j.1469-0705.2001.00335.x. [DOI] [PubMed] [Google Scholar]
- 7.Brown S, Gersen S, Anyane-Yeboa K, Warburton D. Preliminary definition of a “critical region” of chromosome 13 in q32: report of 14 cases with 13q deletions and review of the literature. Am J Med Genet. 1993;45:52. doi: 10.1002/ajmg.1320450115. [DOI] [PubMed] [Google Scholar]
- 8.Bamforth JS, Lin CC. DK phocomelia phenotype (von Voss-Cherstvoy syndrome) caused by somatic mosaicism for del (13q) Am J Med Genet. 1997;73:408. [PubMed] [Google Scholar]
- 9.Iafolla AK, McConkie-Rosell A, Chen YT. VATER and hydrocephalus: distinct syndrome? Am J Med Genet. 1991;38:46. doi: 10.1002/ajmg.1320380112. [DOI] [PubMed] [Google Scholar]
- 10.Paulozzi LJ. International trends in rates of hypospadias and cryptorchidism. Environ Health Perspect. 1999;107:297. doi: 10.1289/ehp.99107297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharpe RM. The ‘oestrogen hypothesis’-where do we stand now? Int J Androl. 2003;26:2. doi: 10.1046/j.1365-2605.2003.00367.x. [DOI] [PubMed] [Google Scholar]
- 12.Myers J, editor. Endocrine Disruption: Emerging Science Vitally Important for Pediatric Urologists. William J. Miller Assoc., Inc.; Pearl River, NY: 2000. pp. 2–3. [Google Scholar]
- 13.Fisher JS. Environmental anti-androgens and male reproductive health: focus on phthalates and testicular dysgenesis syndrome. Reproduction. 2004;127:305. doi: 10.1530/rep.1.00025. [DOI] [PubMed] [Google Scholar]
- 14.Paulozzi LJ, Erickson JD, Jackson RJ. Hypospadias trends in two US surveillance systems. Pediatrics. 1997;100:831. doi: 10.1542/peds.100.5.831. [DOI] [PubMed] [Google Scholar]
- 15.Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 16.Brown SA, Warburton D, Brown LY, Yu C.-y., Roeder ER, Stengel-Rutkowski S, et al. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- 17.Walsh LE, Vance GH, Weaver DD. Distal 13q deletion syndrome and the VACTERL association: case report, literature review, and possible implications. Am J Med Genet. 2001;98:137. doi: 10.1002/1096-8628(20010115)98:2<137::aid-ajmg1022>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 18.Vittu PG, Croquette MF, Donney A, Duminy FR, Couturier J, Cousin J. Syndrome polymalformatif lethal avec deletion 13q secondaire a une translocation maternelle X [in French].13. J Genet Hum. 1989;37:141. [PubMed] [Google Scholar]
- 19.Brock W, Pena A. Urological implications of imperforate anus. AUA Update Series. 1991:202. [Google Scholar]
- 20.Metts JC, 3rd, Kotkin L, Kasper S, Shyr Y, Adams MC, Brock JW., 3rd Genital malformations and coexistent urinary tract or spinal anomalies in patients with imperforate anus. J Urol. 1997;158:1298. doi: 10.1097/00005392-199709000-00168. [DOI] [PubMed] [Google Scholar]
- 21.Lerone M, Bolino A, Martucciello G. The genetics of anorectal malformations: a complex matter. Semin Pediatr Surg. 1997;6:170. [PubMed] [Google Scholar]
- 22.Chung JL, Choi JR, Park MS, Choi SH. A case of del(13) (q22) with multiple major congenital anomalies, imperforate anus and penoscrotal transposition. Yonsei Med J. 2001;42:558. doi: 10.3349/ymj.2001.42.5.558. [DOI] [PubMed] [Google Scholar]
- 23.Boduroglu K, Alikasifoglu M, Tuncbilek E, Uludogan S. Ring chromosome 13 in an infant with multiple congenital anomalies and penoscrotal transposition. Clinical Dysmorphology. 1998;7:299. doi: 10.1097/00019605-199810000-00012. [DOI] [PubMed] [Google Scholar]
- 24.Gershoni-Baruch R, Zekaria D. Deletion (13) (q22) with multiple congenital anomalies, hydranencephaly and penoscrotal transposition. Clinical Dysmorphology. 1996;5:289. [PubMed] [Google Scholar]
- 25.Fryns JP, Peeters R, Petit P, Van den Berghe H. New Chromosomal Syndromes. Acta Paediatr Belg. 1980;33:261. [PubMed] [Google Scholar]
- 26.Parida SK, Hall BD, Barton L, Fujimoto A. Penoscrotal transposition and associated anomalies: report of five new cases and review of the literature. Am J Med Genet. 1995;59:68. doi: 10.1002/ajmg.1320590115. [DOI] [PubMed] [Google Scholar]
- 27.Christensen K, Madsen CM, Hauge M, Kock K. An epidemiological study of congenital anorectal malformations: 15 Danish birth cohorts followed for 7 years. Paediatr Perinat Epidemiol. 1990;4:269. doi: 10.1111/j.1365-3016.1990.tb00650.x. [DOI] [PubMed] [Google Scholar]
- 28.Kiesewetter WB, Chang JH. Imperforate Anus: a five to thirty year follow-up perspective. Prog Pediatr Surg. 1977;10:111. [PubMed] [Google Scholar]
- 29.Spouge D, Baird PA. Imperforate anus in 700,000 consecutive liveborn infants. Am J Med Genet Suppl. 1986;2:151. doi: 10.1002/ajmg.1320250619. [DOI] [PubMed] [Google Scholar]
- 30.Smith E. Incidence, frequency of types and etiology of anorectal malformations. In: Stephens F, Smith E, editors. Anorectal Malformations in Children. Liss; New York, NY: 1988. pp. 231–247. [PubMed] [Google Scholar]
- 31.Perriton CL, Powles N, Chiang C, Maconochie MK, Cohn MJ. Sonic hedgehog signaling from the urethral epithelium controls external genital development. Dev Biol. 2002;247:26. doi: 10.1006/dbio.2002.0668. [DOI] [PubMed] [Google Scholar]
- 32.Haraguchi R, Mo R, Hui C, Motoyama J, Makino S, Shiroishi T, et al. Unique functions of Sonic hedgehog signaling during external genitalia development. Development. 2001;128:4241. doi: 10.1242/dev.128.21.4241. [DOI] [PubMed] [Google Scholar]
- 33.Haraguchi R, Suzuki K, Murakami R, Sakai M, Kamikawa M, Kengaku M, et al. Molecular analysis of external genitalia formation: the role of fibroblast growth factor (Fgf) genes during genital tubercle formation. Development. 2000;127:2471. doi: 10.1242/dev.127.11.2471. [DOI] [PubMed] [Google Scholar]
- 34.Morgan EA, Nguyen SB, Scott V, Stadler HS. Loss of Bmp7 and Fgf8 signaling in Hoxa13-mutant mice causes hypospadia. Development. 2003;130:3095. doi: 10.1242/dev.00530. [DOI] [PubMed] [Google Scholar]
- 35.de Santa Barbara P, Roberts DJ. Tail gut endoderm and gut/genitourinary/tail development: a new tissue-specific role for Hoxa13. Development. 2002;129:551. doi: 10.1242/dev.129.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Post LC, Innis JW. Infertility in adult hypodactyly mice is associated with hypoplasia of distal reproductive structures. Biol Reprod. 1999;61:1402. doi: 10.1095/biolreprod61.6.1402. [DOI] [PubMed] [Google Scholar]
- 37.Warot X, Fromental-Ramain C, Fraulob V, Chambon P, Dolle P. Gene dosage-dependent effects of the Hoxa-13 and Hoxd-13 mutations on morphogenesis of the terminal parts of the digestive and urogenital tracts. Development. 1997;124:4781. doi: 10.1242/dev.124.23.4781. [DOI] [PubMed] [Google Scholar]
- 38.Ogino Y, Suzuki K, Haraguchi R, Satoh Y, Dolle P, Yamada G. External genitalia formation: role of fibroblast growth factor, retinoic acid signaling, and distal urethral epithelium. Ann N Y Acad Sci. 2001;948:13. [PubMed] [Google Scholar]
- 39.Suzuki K, Ogino Y, Murakami R, Satoh Y, Bachiller D, Yamada G. Embryonic development of mouse external genitalia: insights into a unique mode of organogenesis. Evol Dev. 2002;4:133. doi: 10.1046/j.1525-142x.2002.01061.x. [DOI] [PubMed] [Google Scholar]
- 40.Kim J, Kim P, Hui CC. The VACTERL association: lessons from the Sonic hedgehog pathway. Clin Genet. 2001;59:306. doi: 10.1034/j.1399-0004.2001.590503.x. [DOI] [PubMed] [Google Scholar]
- 41.Mo R, Kim JH, Zhang J, Chiang C, Hui CC, Kim PC. Anorectal malformations caused by defects in sonic hedgehog signaling. Am J Pathol. 2001;159:765. doi: 10.1016/S0002-9440(10)61747-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramalho-Santos M, Melton DA, McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development. 2000;127:2763. doi: 10.1242/dev.127.12.2763. [DOI] [PubMed] [Google Scholar]
- 43.Arsic D, Qi BQ, Beasley SW. Hedgehog in the human: a possible explanation for the VATER association. J Paediatr Child Health. 2002;38:117. doi: 10.1046/j.1440-1754.2002.00813.x. [DOI] [PubMed] [Google Scholar]
- 44.Bose J, Grotewold L, Ruther U. Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum Mol Genet. 2002;11:1129. doi: 10.1093/hmg/11.9.1129. [DOI] [PubMed] [Google Scholar]
- 45.Kimmel SG, Mo R, Hui CC, Kim PC. New mouse models of congenital anorectal malformations. J Pediatr Surg. 2000;35:227. doi: 10.1016/s0022-3468(00)90014-9. [DOI] [PubMed] [Google Scholar]
- 46.Dravis C, Yokoyama N, Chumley MJ, Cowan CA, Silvany RE, Shay J, et al. Bidirectional signaling mediated by ephrin-B2 and EphB2 controls urorectal development. Dev Biol. 2004;271:272. doi: 10.1016/j.ydbio.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 47.Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93:741. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- 48.Russell L. Chromosome 13, Monosomy 13q. In: Buyse M, editor. Birth Defects Encyclopedia. Center for Birth Defects Information Services, Inc.; Dover, MA: 1989. pp. 366–367. [Google Scholar]