

Abstract
Atherogenic response to dietary fat and cholesterol challenge was evaluated in mice lacking both the LDL receptor (LDLr−/−) and apoA-I (apoA-I−/−) gene, LDLr−/−/apoA-I−/− or double-knockout mice. Gender- and age-matched LDLr−/−/apoA-I−/− mice were fed a diet consisting of 0.1% cholesterol and 10% palm oil for 16 weeks and compared to LDLr−/− mice or single-knockout mice. The LDLr−/− mice showed a 6- to 7-fold increase in total plasma cholesterol (TPC) compared to their chow-fed mice counterparts, while LDLr−/−/apoA-I−/− mice showed only a 2- to 3-fold increase in TPC compared to their chow-fed controls. This differential response to the atherogenic diet was unanticipated, since chow-fed LDLr−/− and LDLr−/−/apoA-I−/− mice began the study with similar LDL levels and differed primarily in their HDL concentration. The 6-fold diet-induced increase in TPC observed in the LDLr−/− mice occurred mainly in VLDL/LDL and not in HDL. Mid-study plasma samples taken after 8 weeks of diet feeding showed that LDLr−/− mice had TPC concentrations approximately 60% of their 16-week level, while the LDLr−/−/apoA-I−/− mice had reached 100% of their 16-week TPC concentration after only 8 weeks of diet. Male LDLr−/− mice showed similar aortic cholesterol levels to male LDLr−/−/apoA-I−/− mice despite a 4-fold higher VLDL/LDL concentration in the LDLr−/− mice. A direct comparison of the severity of aortic atherosclerosis between female LDLr−/− and LDLr−/−/apoA-I−/− mice was compromised due to the loss of female LDLr−/−/apoA-I−/− mice between 10 and 14 weeks into the study. Diet-fed female and, with time, male LDLr−/−/apoA-I−/− mice suffered from severe ulcerated cutaneous xanthomatosis. This condition, combined with a complete depletion of adrenal cholesterol, manifested in fatal wasting of the affected mice. In conclusion, LDLr−/− and LDLr−/−/apoA-I−/− mice showed dramatic TPC differences in response to dietary fat and cholesterol challenge, while despite these differences both genotypes accumulated similar levels of aortic cholesterol.
The plasma concentration of high-density lipoproteins (HDL) and apolipoprotein A-I (apoA-I) are considered to be reliable measures of an individual’s risk of developing coronary heart disease (CHD). 1 Analysis of four of the largest human epidemiology studies suggest that for every 1 mg/dl increase in HDL apoA-I, a 2% decrease in CHD risk for men and a 3% decrease for women may result. 2 Additionally, the use of animal models has established that inhibition or regression of atherosclerosis is highly dependent on HDL apoA-I concentrations, 3 demonstrated by the use of transgenic mice and rabbits, 4-6 and by repeated intravenous administration of apoA-I. 7-9
A more complicated picture of the role of HDL apoA-I in CHD prevention has emerged from studies in mice with targeted deletions of the mouse apoA-I gene, 10,11 because the mere absence of plasma apoA-I does not lead to a greater atherosclerosis susceptibility in mice. Rather, it appears from studies of transgenic mice with targeted deletions, 12-14 or from studies of somatic gene transfer 15-19 and from studies using combinations of targeted disruptions, as in apoE and apoA-I knockout mice, 20,21 the development of atherosclerosis depends on the type and amount of atherogenic particles in plasma as well as the concentration of HDL apoA-I. Thus, it appears that HDL apoA-I protects the vascular wall in situations in which concentrations of VLDL and LDL could potentially cause development of atherosclerosis. Although this conclusion has not been completely supported by studies in humans who either lack plasma apoA-I or possess apoA-I mutations that lead to very low plasma concentrations of HDL apoA-I, 22 it is likely that populations of individuals with apoA-I mutations are too small to provide reliable statistical analysis.
Therefore, we undertook the present studies to determine the protective effect of HDL apoA-I concentration in the presence of VLDL and LDL concentrations resembling that seen in LDL receptor deficiency in humans. LDL receptor deficient mice 23,24 and apoA-I deficient mice 10,11 were crossed to obtain double-knockout mice, LDLr−/−/apoA-I−/−. These mice were fed a chow or an atherogenic diet and the effects on plasma lipoproteins and atherosclerosis were examined.
Experimental Procedures
Knockout Mice and Experimental Diet
LDL receptor knockout (LDLr−/−) mice and apoA-I knockout (apoA-I−/−) were obtained from Jackson Lab (Bar Harbor, ME) where both lines had been bred six to eight times into the C57BL/6 background. LDLr−/− mice were fully crossed into apoA-I−/− mice, to obtain LDLr−/−/apoA-I−/− mice. All mice were housed at the Wake Forest University Baptist Medical Center where procedures were approved by the Animal Care and Use Committee of the Wake Forest University Health Sciences.
Experimental mice were weaned at 21 days and fed a chow diet (Purina). At 30 days of age blood was obtained by orbital sinus bleeding after a 4- to 6-hour fast for baseline evaluation. The animals were anesthetized using a 1:1 mixture of ketamine (50 mg/ml) and xylazine (10 mg/ml) in which 1 μl per gram body weight was injected intramuscularly behind the knee. The orbital sinus bleeding was performed by inserting a micro hematocrit capillary tube into the retro-orbital plexus. Up to 250 μl of whole blood was drawn. The animal’s eye was treated with an opthalamic ointment (Lacri-Lube) to keep the eyes lubricated and the mouse was then placed on a 37°C heating pad. The respiration, orientation, and balance of the mouse was monitored for at least 2 hours. At 6 weeks of age, a group of mice were fed an atherogenic diet containing 10% saturated fat from palm oil and 0.1% cholesterol for 16 weeks, as previously described. 25,26 As controls, both gender- and age-matched mice of each genotype were fed a Purina chow diet for 16 weeks. All mice were maintained in a temperature-controlled room with a 12-hour light and 12-hour dark cycle.
At the end of the diet period both chow- and diet-fed mice were fasted then anesthetized as described above. Blood was obtained by cardiac puncture. Approximately 1 ml of blood was collected in a 1.5-ml tube containing 20 μl of 0.5 μmol/L ethylenediaminetetraacetate (EDTA) pH 8.0/1 μmol/L sodium azide and placed immediately on ice. The blood was centrifuged immediately at 4°C at 10,000 rpm for 10 minutes. The plasma was removed and stored at −20°C. The animal was opened via midline laparotomy to expose the thoracic and abdominal cavities. The heart and aorta were removed and suspended in 10% formalin for at least 48 hours before further analysis. For all other tissues, sections were cut and stored in 10% formalin. For chemical analysis additional sections of tissue were frozen in liquid nitrogen and stored at −80°C for later use.
Plasma Lipoprotein Analysis
Plasma from both chow- and diet-fed mice was analyzed for total plasma, free and esterified cholesterol and triglycerides by enzymatic assay (Roche Molecular Biochemical, Indianapolis, IN). HDL cholesterol was determined after dextran-sulfate precipitation of plasma as described previously. 27,28
The plasma lipoprotein distribution was determined for chow- and diet-fed mice using fast protein liquid chromatography (FPLC). Approximately 200 μl of plasma from chow-fed and 100 μl from diet-fed mice were applied to two Superose-6 columns connected in tandem and run at 0.5 ml/min in a buffer containing 0.9% NaCl, 0.26 mmol/L EDTA (pH 7.4), and 1.5 mmol/L NaN3. Approximately 75,500-μl fractions were collected and 50 to 100 μl from each fraction was used for lipid determination, as previously described. 28 To assess the apoprotein distribution, 10 μl from chow or 20 μl from diet-fed FPLC-fractionated plasma were separated on a 13% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), electrotransferred to nitrocelluose at 4°C for 1 hour at 1 Amp using 25 mmol/L Tris pH 8.0 192 mmol/L glycine and 20% methanol. Western analysis was carried as previously described 28 using antibodies raised against mouse apoA-I or mouse apoE (Biodesign, Saco, ME).
Analysis of Plasma Apoprotein Distribution
To assess the apoprotein distribution in mouse plasma from LDLr−/− and LDLr−/−/apoA-I−/− mice, freshly drawn plasma was subjected to density-gradient ultracentrifugation. Approximately 250 μl of plasma was adjusted to 1.27 g/ml using KBr, then overlayered with 12 to 13 ml of a 1.25 g/ml KBr solution. The tubes were sealed, placed in a pre-cooled Ti 70.1 rotor and centrifuged at 4°C, 50,000 rpm for 18 hours. The top 3 ml from each centrifuge tube was exhaustively dialyzed against 10 mmol/L ammonium bicarbonate (pH 7.4), 3 μmol/L EDTA, and 15 μmol/L sodium azide. Following dialysis, the protein concentration was determined by Lowry assay. 29 Aliquots containing 10 μg of protein were concentrated and separated by SDS-PAGE on 4% to 30% gradient gels. Each lane contained approximately 10 μg of protein and was compared to the Mark 12 molecular weight standard (Invitrogen, Carlsbad, CA). Gels were stained with Coomassie brilliant blue, destained and their image recorded using an α Innotech Imaging system.
Atherosclerosis and Tissue Cholesterol Measurements
Atherosclerosis evaluations were carried out as previously described. 25,26 Briefly, the heart and aorta were placed under a dissecting microscope and the adventitia was completely removed. Then the aorta was detached at the base of the heart, blotted, weighed and placed in a glass tube with 3 ml of chloroform-methanol 2:1 (v/v), and 5α-cholestane as the internal standard. Extracted lipid was dried under a stream of N2 at 60°C to remove solvent. The lipid was dissolved in hexane and injected onto a ZB-50 gas-liquid chromatography (GLC) column for free cholesterol determination. The remaining lipid extract was dried again, dissolved in 1 ml of ethanol and 0.1 ml 50% KOH was added to each tube. The mixture was then heated at 60°C for 1 hour. Following the incubation, 0.5 ml of hexane and 1 ml of water was added, the tubes were centrifuged to separate the phases. The top phase was transferred and injected onto a DB17 GLC column for total cholesterol measurement. Esterified cholesterol was calculated as the difference between free and total cholesterol. Finally, delipidated aortic tissue was digested and dissolved in 1N NaOH and the total protein content determined by Lowry. 29
Adrenal glands from both chow- and diet-fed mice were taken at the time of necropsy, weighed and then placed under a dissecting microscope. The surrounding fat was removed before GLC analysis using the same method as described above for the quantification of cholesterol in the mouse aorta. 25,26 Sections of skin were taken from the back and abdomen at the time of necropsy and frozen. At the time of analysis, 200 to 400 mg of tissue were subjected to lipid extraction and aliquots were taken, dried, and re-suspended in water and 0.5% Triton-X100. Enzymatic analysis was performed on aliquots to determine free and total cholesterol content. All samples were compared to cholesterol standards of known concentration, which had been treated in an analogous manner.
Histology
Sections of liver, skin, and adrenal gland from chow- and diet-fed mice were fixed with 10% formalin at the end of the study. These tissues were embedded in paraffin, sectioned, and then stained with eosin and counterstained with hematoxylin. The slides were examined under the Zeiss Axioplan 2 microscope and digital images recorded. Images were then prepared for publication using Adobe Photoshop 6.0.
Data Analysis
Data are presented as the mean ± SEM or ± SD as indicated. Data were analyzed by one-way analysis of variance using StatView 5.0.1 and then individual differences between pairing of groups were found using Fisher’s least significant post hoc test. Statistical significance was considered at P ≤ 0.05.
Results
Time-Course Effects of Diet on Plasma Lipid Levels
This study was initiated to determine the effects of a dietary fat and cholesterol challenge on LDLr−/−/apoA-I−/− mice. All mice were age- and gender-matched and fed either chow or an atherogenic diet, comprised of 0.1% cholesterol and 10% palm oil, for 16 weeks. After 8 weeks of consuming the atherogenic diet the mice were bled, and plasma lipid determinations were conducted. After 16 weeks of diet-feeding, all mice were euthanized and complete plasma lipoprotein and atherosclerosis analyses were conducted. Table 1 ▶ shows the plasma lipid values for chow- and diet-fed LDLr−/− or single-knockout mice and LDLr−/−/apoA-I−/− or double-knockout mice after 8 and 16 weeks of diet-feeding. Chow-fed LDLr−/− and LDLr−/−/apoA-I−/− mice showed distinctly different TPC values which ranged between 200 to 350 mg/dl, with no significant differences between males and females within a genotype. After consuming the atherogenic diet for 8 weeks the LDLr−/− mice showed TPC levels that were approximately 60% to 70% of their 16-week level, reaching 1482 ± 166 mg/dl, while after 8 weeks of consuming the diet, LDLr−/−/apoA-I−/− mice reached their maximum TPC concentration of 700 ± 37 mg/dl (Table 1) ▶ . The significant difference between the two genotypes was that LDLr−/− mice showed a 7-fold increase in TPC compared to their chow-fed counterparts following 16 weeks of diet, while LDLr−/−/apoA-I−/− mice showed only a modest 3-fold increase in TPC after 16 weeks of diet, with neither genotype showing gender-related differences (Table 1) ▶ .
Table 1.
Plasma Lipid Levels in LDLr−/− and LDLr−/−/Apo A-I−/− Mice
| Genotype (n) | Gender | TPC (mg/dl) | FC (mg/dl) | EC (mg/dl) | TG (mg/dl) | FC/TC |
|---|---|---|---|---|---|---|
| 16 weeks of chow | ||||||
| LDLr−/− (5) | Female | 370 ± 43a | 98 ± 10a | 271 ± 32a | 75 ± 4a | 0.31 ± 0.02a |
| LDLr−/− (4) | Male | 302 ± 18a | 82 ± 22a | 220 ± 17a | 109 ± 13b | 0.32 ± 0.01a |
| LDLr−/−/apo A–I−/− (5) | Female | 192 ± 15b | 74 ± 16a | 118 ± 15b | 101 ± 16b | 0.45 ± 0.03b |
| LDLr−/−/apo A–I−/− (5) | Male | 212 ± 24b | 80 ± 12a | 127 ± 28b | 152 ± 11c | 0.47 ± 0.03b |
| 8 weeks of diet | ||||||
| LDLr−/− (5) | Female | 1577 ± 121c | 444 ± 27b | 1133 ± 113c | 202 ± 39c | 0.29 ± 0.022c |
| LDLr−/− (4) | Male | 1388 ± 211c | 376 ± 42b | 1011 ± 171c | 109 ± 24b | 0.28 ± 0.020c |
| LDLr−/−/apo A–I−/− (7) | Female | 630 ± 33d | 202 ± 6c | 427 ± 28d | 90 ± 17a,b | 0.36 ± 0.012d |
| LDLr−/−/apo A–I−/− (17) | Male | 748 ± 42d | 245 ± 12c | 488 ± 39d | 109 ± 24b | 0.37 ± 0.009d |
| 16 weeks of diet | ||||||
| LDLr−/− (5) | Female | 2341 ± 112c | 555 ± 17d | 1768 ± 95e | 214 ± 18c | 0.24 ± 0.008e |
| LDLr−/− (4) | Male | 2226 ± 108e | 520 ± 20d | 1705 ± 9e | 136 ± 15c | 0.23 ± 0.005e |
| LDLr−/−/apo A–I−/− (7) | Female | 546 ± 50d | 187 ± 13c | 359 ± 30d | 264 ± 19d | 0.37 ± 0.001d |
| LDLr−/−/apo A–I−/− (17) | Male | 652 ± 22d | 197 ± 15c | 456 ± 20d | 188 ± 15f | 0.33 ± 0.009a |
Blood samples were collected after either a 4- to 6-hour fast. Plasma lipid values were determined by enzymatic assay as described in Experimental Procedures. All values represent the mean ± SEM, n = number of animals in the indicated group. Lowercase letters indicate significant differences at P < 0.05.
In contrast to plasma cholesterol levels, 8 weeks triglyceride values for LDLr−/− mice were 155 ± 29 mg/dl and increased slightly to 200 ± 15 mg/d by 16 weeks (Table 1) ▶ , while triglycerides in LDLr−/−/apoA-I−/− mice steadily increased from their 8-week value of 98 ± 20 mg/dl to 220 ± 17 mg/dl by 16 weeks (Table 1) ▶ . At 16 weeks of diet, female LDLr−/−/apoA-I−/− mice showed a 1.4-fold higher triglyceride level than male LDLr−/−/apoA-I−/− mice (Table 1) ▶ .
Both the type of diet and duration of the feeding affected the plasma ratio of free and total cholesterol in LDLr−/− and LDLr−/−/apoA-I−/− mice in this study. Chow-fed LDLr−/− mice had an FC/TC ratio of 0.31 ± 0.02, while LDLr−/−/apoA-I−/− mice averaged a FC/TC ratio of 0.46 ± 0.03. The increase in FC relative to TC strongly suggests that the absence of plasma apoA-I in the LDLr−/−/apoA-I−/− mice resulted in impairment of LCAT-mediated cholesterol esterification. Reduced LCAT activation yields less plasma ester cholesterol (EC) and the reduction in plasma EC is not apparently overcome by the presence of other plasma apoproteins that might activate LCAT (apoE, apoAIV, and apoCs), suggesting that in mice, apoA-I assumes a major role in regulating plasma EC concentrations. 22,28 Supporting this idea, a report in apoA-I−/− mice 10,30 showed that the relative increase in the free cholesterol/total cholesterol (FC/TC) ratio or the concomitant decrease in the EC/TC ratio is associated with a lack of plasma apoA-I, since control mice expressing both apoA-I+/+ alleles displayed an EC/TC ratio of 0.60, and mice lacking both apoA-I−/− alleles showed an EC/TC ratio of 0.38.
The FC/TC ratio was also affected by the length of time the mice were fed an atherogenic diet. When mice of either genotype were fed for 8 weeks, the FC/TC ratio was significantly reduced from the chow-fed control level, as seen in Table 1 ▶ . This trend continued, and by 16 weeks the FC/TC ratio had dropped approximately 28% in LDLr−/− mice and by 24% in LDLr−/−/apoA-I−/− mice, when compared to their chow-fed counterparts. It is possible that this decrease reflects an accumulation of VLDL and LDL CE since these particles are not cleared via the LDL receptor 23,31 in these mice.
Plasma Lipoprotein and Apolipoprotein Distribution
Chow-fed LDLr−/− and LDLr−/−/apoA-I−/− mice averaged TPC values of 336 ± 30 and 202 ± 20 mg/dl, respectively. A portion of this variance in TPC levels between the two genotypes of mice was due to differences in HDL cholesterol concentration, as shown in Figure 1A ▶ . An approximate 50 mg/dl difference was seen between mice expressing and mice lacking endogenous apoA-I, as previously shown in apoA-I−/− mice. 10,28,30 After 16 weeks of consuming the atherogenic diet, HDL cholesterol levels were not significantly different from their chow-fed counterparts for either genotype, as shown in Figure 1B ▶ .
Figure 1.
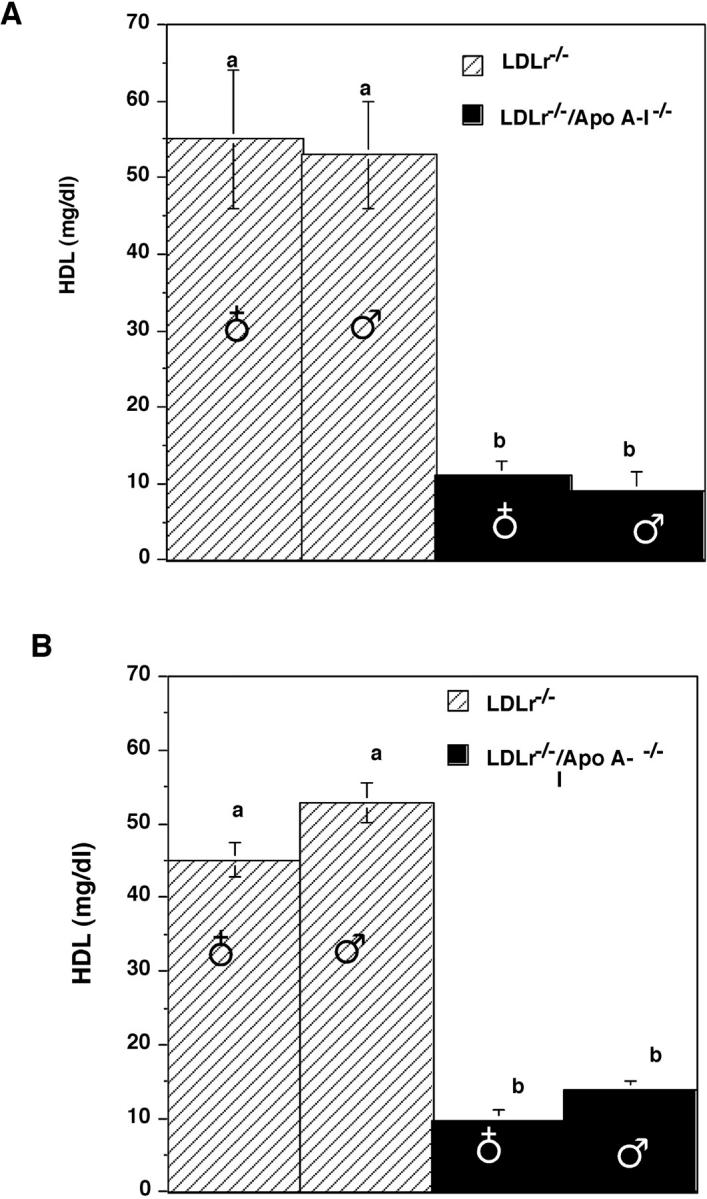
A: HDL cholesterol concentration in chow-fed LDLr−/− mice (striped bar) and LDLr−/−/apoA-I−/− mice (solid bar). B: Sixteen-week diet-fed LDLr−/− mice (striped bar) and LDLr−/−/apoA-I−/− mice (solid bar). HDL cholesterol was determined as described in Experimental Procedures. All values represent the mean ± SEM. Values with unlike superscripts indicate significant differences at P < 0.01.
The plasma lipoprotein cholesterol and triglyceride distributions were characterized after FPLC separation of whole mouse plasma. Figure 2, A and B ▶ show the total cholesterol distribution for chow- and diet-fed LDLr−/− and LDLr−/−/apoA-I−/− mice, respectively, and total triglyceride distribution (Figure 2, C and D) ▶ for chow- and diet-fed LDLr−/− and LDLr−/−/apoA-I−/− mice, respectively. Both LDLr−/− and LDLr−/−/apoA-I−/− mice had similar LDL cholesterol levels as shown in Figure 3A ▶ , but differed predominantly in their HDL cholesterol content.
Figure 2.

FPLC separation of plasma lipoprotein showing the distribution of total cholesterol (A and B) and triglyceride (C and D) in LDLr−/− (open square) and LDLr−/−/apoA-I−/− mice (filled circle) fed chow (A and C) or a diet containing 0.1% cholesterol and 10% palm oil for 16 weeks (B and D). Approximately 200 μl of chow-fed mouse plasma or 100 μl of diet-fed mouse plasma was applied to two Superose-6 columns connected in tandem and run at 0.5 ml/min, as described in Experimental Procedures. Total cholesterol and triglyceride concentrations were measured in each individual fraction by enzymatic assay. Western blot analysis was performed on each FPLC fraction using 10 μl from chow-fed plasma fractions and 20 μl from diet-fed plasma fractions as described in Experimental Procedures.
Figure 3.

Coomassie-blue-stained 4% to 30% SDS-PAGE of the d < 1.25 g/ml fraction from mouse plasma. Lanes 1 and 2 show the d <1.25 g/ml tops from two different chow-fed LDLr−/−/apoA-I−/−mice; lanes 3 and 4 show the d < 1.25 g/ml tops from two different chow-fed LDLr−/− mice; lanes 5 and 6 show the d < 1.25 g/ml tops from two different chow-fed LDLr−/− mice; lanes 7 and 8 show the d < 1.25 g/ml tops from two different diet-fed LDLr−/−/apoA-I−/− mice. The d < 1.25 g/ml plasma fraction was isolated by density ultracentrifugation of 100 to 200 μl of mouse plasma as described in Experimental Procedures. Each lane is the equivalent of 10 μg of total d < 1.25 g/ml lipoprotein. The gel was stained/destained then photographed using an α Innotech Imager.
After 16 weeks of consuming an atherogenic diet both LDLr−/− and LDLr−/−/apoA-I−/− mice showed a large increase in VLDL and LDL total cholesterol as shown in Figure 2B ▶ . However, the LDLr−/− mice showed an approximate 6-fold increase relative to their chow-fed counterparts, while in LDLr−/−/apoA-I−/− mice, the VLDL/LDL levels increased by only 3-fold relative to chow-fed LDLr−/−/apoA-I−/− mice.
The triglyceride distribution in chow- and diet-fed LDLr−/− mice was similar to LDLr−/−/apoA-I−/− mice (Figure 2, C and D) ▶ , except that LDLr−/−/apoA-I−/− mice consistently showed a 30% to 40% higher level of triglyceride under both dietary conditions (Table 1) ▶ . On the other hand, the apoE distribution was strikingly different between LDLr−/− and LDLr−/−/apoA-I−/−, as shown by Western analysis of the fractional apoE distribution as shown below the FPLC profile. These results suggested that the abundance of mouse apoE on VLDL- and LDL-sized particles in LDLr−/−/apoA-I−/− mice may be due to the reduced size and amount of HDL apoA-I particles in these mice, since in LDLr−/− mice, lipid-bound apoE was more equally distributed between LDL- and HDL-sized particles.
Changes in plasma apoprotein levels in response to diet were assessed by comparing the d <1.25 g/ml fraction from plasma of chow- and diet-fed LDLr−/− and LDLr−/−/apoA-I−/− mice. Figure 3 ▶ shows a Coomassie-blue-stained 4% to 30% SDS-gradient gel of the d <1.25 g/ml from chow- and diet-fed LDLr−/− and LDLr−/−/apoA-I−/− mice. In chow-fed LDLr−/−/apoA-I−/− mice the major apoproteins present were apoB-100 and apoE, with the obvious exception of apoA-I, while in LDLr−/− mice, the predominant apoproteins were apo B-100, apoA-I, and apoE. As seen in previous studies on apoA-I−/− knockout mice, 11 chow-fed LDLr−/−/apoA-I−/−mice had levels of apoE approximately 3-fold to 4-fold higher than the apoE levels in LDLr−/− mice (Figure 3) ▶ . In response to diet, the apo B-48 and apoE levels in both LDLr−/− and LDLr−/−/apoA-I−/− mice were elevated, with LDLr−/− mice showing a 5-fold to 6-fold increase in apoE and a 2-fold lower level of apoA-I. While in LDLr−/−/apoA-I−/− mice the atherogenic diet induced a 3-fold higher level of apoE when compared to chow-fed LDLr−/−/apoA-I−/− mice. Overall, the diet-fed LDLr−/−/apoA-I−/− mice retained a 1.5- to 2-fold higher apoE concentration than diet-fed LDLr−/− mice.
Atherosclerosis Evaluations
The dramatic diet-induced difference in plasma lipoprotein response between single- and double-knockout mice was unexpected considering the similarity in TPC between the two phenotypes when fed chow, the differences relating mainly to HDL concentrations. To assess the major indicator of the effect of diet on aortic cholesterol deposition, atherosclerosis evaluations were performed. The amount of free and ester cholesterol were quantified from whole mouse aorta as previously described 25,26,32 and the values are shown in Table 2 ▶ . The total aortic cholesterol was highest in female LDLr−/− mice but was not statistically different from male LDLr−/− and male LDLr−/−/apoA-I−/−mice. Only female LDLr−/−/apoA-I−/− mice showed statistically lower aortic cholesterol levels (Table 2) ▶ . This trend was also seen when comparing esterified cholesterol values, which showed that female and male LDLr−/− mice had statistically higher aortic esterified cholesterol than female or male LDLr−/−/apoA-I−/− mice. These results strongly suggest that despite the 6-fold to 7-fold higher TPC in diet-fed LDLr−/− mice compared to LDLr−/−/apoA-I−/− mice, the atherogenic potential of plasma lipoprotein in diet-fed LDLr−/−/apoA-I−/− is as severe in driving cholesterol deposition.
Table 2.
Total Aortic Cholesterol Content from LDLr−/− and LDLr−/−/Apo A–I−/− Mice Fed an Atherogenic Diet for 16 Weeks
| Genotype (n) | Gender | TC (μg/mg protein) | FC (μg/mg protein) | EC (μg/mg protein) |
|---|---|---|---|---|
| LDLr−/− (5) | Female | 106.2 ± 3.4a | 52.4 ± 1.8 | 53.7 ± 2.3a |
| LDLr−/− (4) | Male | 81.9 ± 9.5a,c | 41.0 ± 4.6 | 40.8 ± 4.9a,c |
| LDLr−/−/apo A–I−/− (7) | Female | 53.9 ± 8.1b,c | 34.3 ± 4.9 | 19.6 ± 3.2b |
| LDLr−/−/apo A–I−/− (17) | Male | 86.6 ± 7.4a | 49.6 ± 3.6 | 36.9 ± 4.0c |
| ANOVA | 0.01 | NS | 0.001 |
All values represent mean ± SEM, n = number of animals in the indicated group. Aortic total and free cholesterol were determined by GLC as described in Experimental Procedures. Lowercase letters indicate significant differences at the indicated P value. ANOVA, analysis of variance; NS, not significant.
Cutaneous Xanthomatosis, Pruritus, and the Role of Gender in LDLr −/−/apoA-I−/− Mice
Diet-fed single- and double-knockout mice were similar in phenotype except for the onset of severe pruritus, observed in female LDLr−/−/apoA-I−/− mice, resulting in their euthanasia before the end of the 16-week diet study. It is likely that the physiological severity of this condition, as well as death of mice during the study, compromised the atherosclerosis endpoint comparison between female LDLr−/− and LDLr−/−/apoA-I−/− mice (Table 2) ▶ . We first noticed that female LDLr−/−/apoA-I−/− mice had developed severe cutaneous lesions after 9 weeks of eating the atherogenic diet (containing 0.1% cholesterol and 10% palm oil) and then as the scratching and lesion area progressed affected animals ceased to eat or drink. Lesions were characterized by severe ulcerations on thickened skin found predominately on the abdomen, neck, and front limbs (Figure 4B) ▶ , compared to the coat of a 16-week diet-fed male LDLr−/−/apoA-I−/− mouse (Figure 4A) ▶ . These ulcerated lesions appeared to be distinct from non-fatal dermal xanthomatosis described in other animal models of hypercholesterolemia such as in LDLr−/−, 24 LDLr−/−/ACAT1−/− and apoE−/−/ACAT1−/− mice, 33,34 since cholesterol deposition in these models 24,33,34 are characterized by partial hair loss or thinning at the site of raised thickened skin, as we also observed in one 16-week diet-fed male LDLr−/− mouse (Figure 4C) ▶ . Skin thickening in this diet-fed LDLr−/− mouse was not associated with open ulcerated lesions or death resulting from diminished eating and drinking.
Figure 4.

Cutaneous xanthomatosis and severe pruritus in diet-fed LDLr−/−/apoA-I−/− and LDLr−/− mice. A: External appearance of a 14-week diet-fed male LDLr−/−/apoA-I−/− mouse, compared to a 14-week diet-fed female LDLr−/−/apoA-I−/− mouse (B). Female mice began showing severe ulcerated lesions on the abdomen, neck, and front limbs after approximately 9 weeks of consuming the atherogenic diet (containing 0.1% cholesterol, 10% palm oil). C: The skin lesions affecting the double-knockout mice were distinct from the hair loss and scaly thickened skin occasionally seen in diet-fed LDLr−/− mice (C). D: Frequency of ulcerated lesions in male and female LDLr−/−/apoA-I−/− mice after 14 weeks.
Skin lesions observed in our > 9-week diet-fed LDLr−/−/ apoA-I−/− mice appeared to be more similar to lesions reported in diet-fed LDLr−/−/ABCA1−/− and apoE−/−/ABCA1−/− mice. 35 In this study, chow-fed apoE−/−/ABCA1−/− and diet-fed LDLr−/−/ABCA1−/− mice showed cutaneous abnormalities at an early age and were characterized by the presence of ulcerations and excoriations leading to pruritus. 35 Quantification of skin cholesterol in apoE−/−/ABCA1−/− mice indicated that free cholesterol increased 2.5-fold and cholesterol esters increased 7.5-fold compared to age- and gender-matched apoE−/− mouse skin, concluding that lipid accumulation and severity of cell infiltrates were independent of gender and correlated more significantly with age. However, our results showed that ulcerated skin lesions were more prevalent in diet-fed female LDLr−/−/apoA-I−/− mice than males, but with time lesions also developed in male LDLr−/−/apoA-I−/− mice, and by 14 weeks of diet-feeding approximately 60% of male LDLr−/−/apoA-I−/− mice had ulcerated lesions as shown by the graph in Figure 4D ▶ .
Histological examinations were performed on matched skin sections taken from male and female LDLr−/− and LDLr−/−/apoA-I−/− mice. The skin of a diet-fed female LDLr−/− mice displayed normal dermal-layer thickness (Figure 5A) ▶ , whereas skin from a diet-fed male LDLr−/− /apoA-I−/− mouse showed a thickened dermal layer, infiltrated with fat-laden macrophages and cholesterol clefts (Figure 5B) ▶ . Skin examined from a female diet-fed LDLr−/− /apoA-I−/− mouse showed an even more dramatic thickening of the dermis, associated with massive macrophage infiltration and cholesterol deposits apparently disrupting the subcutaneous adipose tissue layer which was not apparent (Figure 5C) ▶ .
Figure 5.
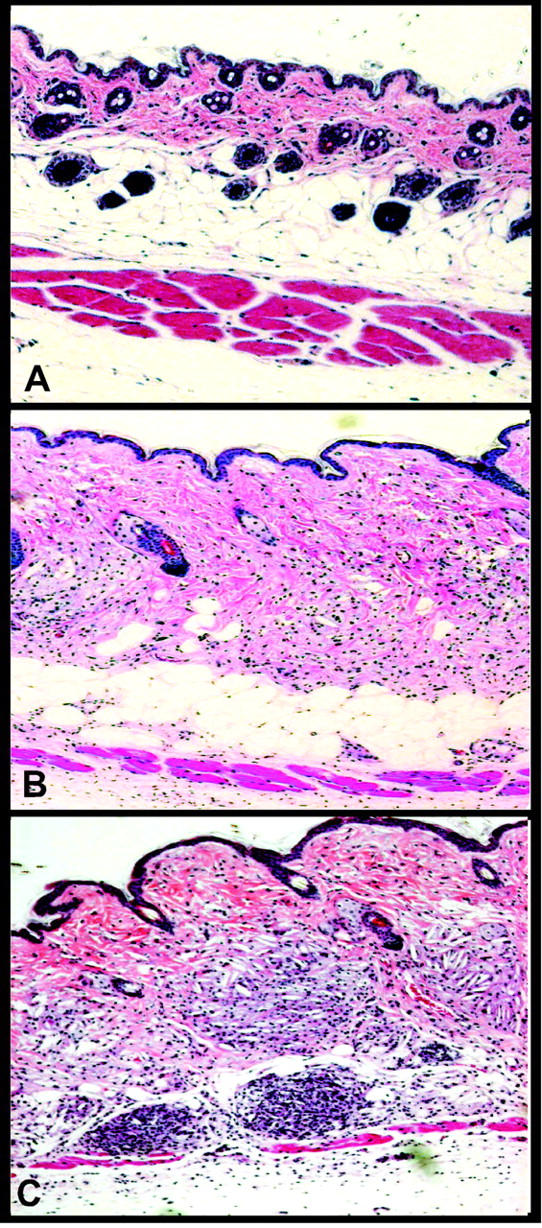
Histopathology of dermal xanthomatosis in LDLr−/−/apoA-I−/− mice. A: H&E-stained section of skin from a 16-week diet-fed LDLr−/− female mouse. B: Skin section from a 16-week diet-fed LDLr−/−/apoA-I−/− male mouse with a thickened dermal layer infiltrated with fat-laden macrophages and cholesterol clefts. C: Skin section from a diet-fed female LDLr−/−/apoA-I−/− mouse showing dramatic thickening of the dermis, associated with massive macrophage infiltration and cholesterol deposits apparently disrupting the subcutaneous adipose tissue layer.
To substantiate our microscopic observations, the free and total cholesterol content was measured on matched skin sections from 16-week diet-fed female (n = 5) and male (n = 5) LDLr−/−/apoA-I−/− mice and compared to diet-fed male (n = 5) and female (n = 5) LDLr−/− mice. The total skin cholesterol content for LDLr−/−/apoA-I−/− mice was 12- to 13-fold higher than for LDLr−/− mice, with no apparent difference between males and females of either genotype. The free cholesterol skin content for LDLr−/− /apoA-I−/− mice was 5.5-fold higher than for LDLr−/− mice (P. Tahiri and M. Sorci-Thomas, unpublished observations). Thus, it appears that LDLr−/−/apoA-I−/− mice accumulate a significantly higher mass of esterified cholesterol within the skin than LDLr−/− mice do in response to the atherogenic diet.
Adrenal Histology and Cholesterol Content in LDLr−/ −/apoA-I−/ − Mice
HDL apoA-I has been shown to be essential for cholesterol delivery to steroidogenic cells in rodents 36-38 therefore, we examined adrenal histology and measured the adrenal cholesterol content in chow and diet-fed LDLr−/− and LDLr−/−/apoA-I−/− mice. Figure 6, A and B ▶ show hematoxylin and eosin (H&E)-stained adrenal sections from chow-fed LDLr−/− male and female mice, and Figure 6, C and D ▶ show sections from 16-week diet-fed LDLr−/− male and female mice, respectively. In each of these panels, abundant cytoplasmic lipid droplets are present in the zona fasiculata, whereas Figure 6, E and F ▶ represent adrenal sections from chow-fed male and female LDLr−/− /apoA-I−/− mice, and Figure 6, G and H ▶ show adrenals from 16-week diet-fed male and female LDLr−/−/apoA-I−/− mice, respectively. In these panels the appearance of the zona fasiculata in both male and female LDLr−/− /apoA-I−/− mice are indicative of severe depletion of cytoplasmic lipid droplets.
Figure 6.
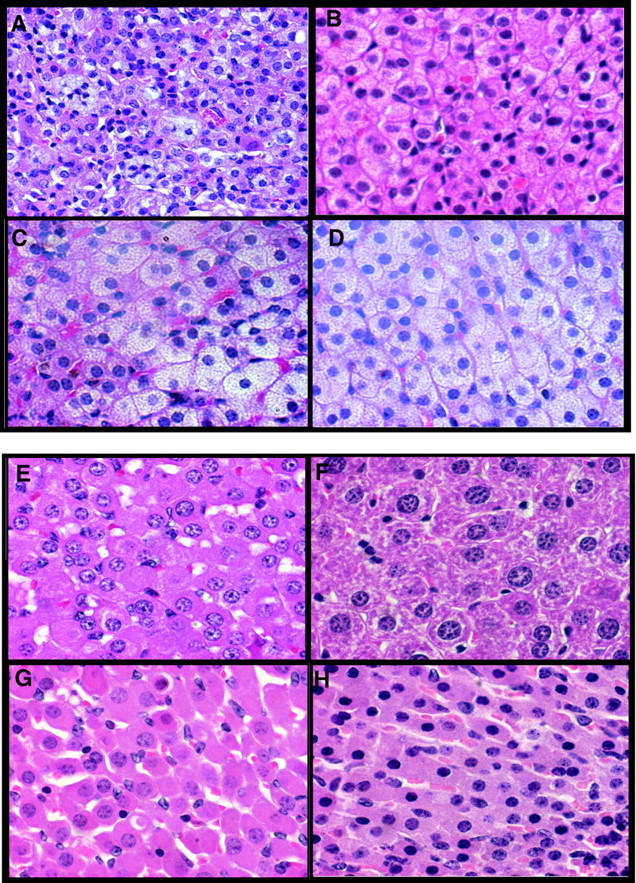
Adrenal sections showing zona fasiculata cells from chow- and diet-fed LDLr−/− and LDLr−/−/apoA-I−/− mice. A and B: Sections from chow-fed male and female LDLr−/− mice, respectively. C and D: Sixteen-week diet-fed male and female LDLr−/− mice, respectively. E and F: Sections from chow-fed male and female LDLr−/−/apoA-I−/− mice, respectively. G and H: Sections from 16-week diet-fed male and female LDLr−/−/apoA-I−/− mice, respectively. Magnification, ×63.
These macroscopic observations are supported by GC measurement of free and esterified cholesterol content, as listed in Table 3 ▶ . Results show that adrenals from female chow-fed LDLr−/− mice had a 5-fold higher total cholesterol content than female LDLr−/−/apoA-I−/− mice. Adrenals from diet-fed female LDLr−/− mice had a 10-fold higher total cholesterol level than female LDLr−/−/apoA-I−/− mice noted. The large decrease was mostly seen in the adrenal esterified cholesterol fraction (Table 3) ▶ since the FC fraction was not statistically affected. Table 3 ▶ also shows that chow-fed female LDLr−/− mice had an approximately 10-fold lower adrenal EC content than female LDLr−/−/apoA-I−/− mice, and that diet-fed LDLr−/−/apoA-I−/− female mice had an approximate 40-fold lower adrenal EC than female LDLr−/− mice. Differences observed between chow- and diet-fed LDLr−/− mice did not reach statistical significance; however, female LDLr−/−/apoA-I−/− mice fed diet for 16 weeks showed a significant reduction in EC when compared to chow-fed female LDLr−/−/apoA-I−/− (Table 3) ▶ . These data support the concept that apoA-I is required for normal cholesterol ester deposits in the adrenal 36 and that depletion of adrenal cholesterol in LDLr−/−/apoA-I−/− mice may contribute to the onset of the fatal diet-related inflammation observed.
Table 3.
Adrenal Cholesterol in LDLr−/− and LDLr−/−/Apo A–I−/− Mice Fed Chow or an Atherogenic Diet for 16 weeks
| Genotype (n) | TC (μg/adrenal) | FC (μg/adrenal) | EC (μg/adrenal) |
|---|---|---|---|
| Chow-Fed | |||
| LDLr−/− (3) | 104.0 ± 42.7a | 20.3 ± 6.9 | 113.6 ± 35.6a |
| LDLr−/−/Apo A–I−/− (2) | 23.8 ± 12.3b | 11.2 ± 3.8 | 12.5 ± 6.4b |
| Diet-Fed | |||
| LDLr−/− (3) | 97.9 ± 25.9a | 12.5 ± 4.8 | 85.4 ± 21.4a |
| LDLr−/−/Apo A–I−/− (4) | 12.2 ± 6.4b | 11.4 ± 8.5 | 1.8 ± 0.2c |
Adrenal total and free cholesterol were determined by GLC as described in Experimental Procedures. All values represent mean ± SD of cholesterol mass per total adrenal as determined in (n) female mice. Lowercase letters indicate significant differences at P < 0.05.
Hepatic Histology and Cholesterol Content in LDLr−/ −/apoA-I−/− Mice
We next examined liver tissue from both male and female LDLr−/− and LDLr−/−/apoA-I−/− mice. Chow-fed LDLr−/− and LDLr−/−/apoA-I−/− mice had similar amounts of hepatic cholesterol which ranged between 4 and 6 mg/g (wet weight) (data not shown), with female mice of either genotype consistently having a lighter organ weight but similar amounts of cholesterol per wet weight of liver (data not shown). Liver triglycerides were similar for chow-fed mice of both genotypes (31 mg/mg wet weight) with no gender differences noted. After 16 weeks of the atherogenic diet, the liver cholesterol content increased for both LDLr−/− and LDLr−/−/apoA-I−/− mice when compared to their chow-fed counterparts, showing an 11-fold increase (52 mg/mg wet weight) for LDLr−/− mice and a 2.5-fold increase (12 mg/mg wet weight) for LDLr−/−/apoA-I−/− mice (data not shown). Liver triglycerides were also increased (3.5-fold) in response to the diet when compared to chow-fed mice, but there were no apparent genotype or gender differences noted.
The higher hepatic cholesterol content in diet-fed LDLr−/− compared to LDLr−/−/apoA-I−/− mice was also seen at the microscopic level. Histological examination revealed liver sections from diet-fed LDLr−/− mice (Figure 7B) ▶ , contained larger diameter and more frequent lipid droplets, compared to chow-fed mice of either genotype (Figure 7A) ▶ . In contrast, the LDLr−/−/apoA-I−/− mice of either gender showed numerous small diameter droplets, shown in Figure 7C ▶ . Histological examination also revealed that in contrast to the single-knockout mice, LDLr−/−/apoA-I−/− mice showed an accumulation of inflammatory cells. Figure 7D ▶ shows a representative section from the liver of a female LDLr−/−/apoA-I−/− mouse characterized by the accumulation of neutrophils and leukocytes around the hepatic vein.
Figure 7.
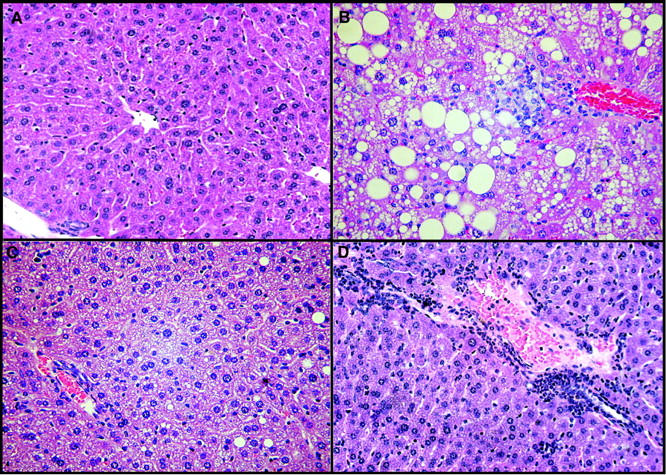
A: H&E-stained liver sections from chow-fed male LDLr−/−/apoA-I−/− mouse. B: A diet-fed male LDLr−/− mouse. C: A diet-fed male LDLr−/−/apoA-I−/− mouse. D: A diet-fed female LDLr−/−/apoA-I−/− mouse. Magnification, ×40.
Discussion
In these studies we have examined the effects of dietary fat and cholesterol on a new double-knockout mouse model in which the genes for both apoA-I and the LDL receptor have been functionally removed. The LDLr−/−/apoA-I−/− or double-knockout mouse showed a number of interesting phenotypes that set them apart from the single-knockout models from which they were generated. One of the most significant was the effect of the atherogenic diet on TPC concentrations. The LDLr−/−/apoA-I−/− mice showed only a 2- to 3-fold increase in TPC which remained relatively constant from 4 weeks to the end of the 16-week study, while the TPC concentration in LDLr−/− mice continued to increase from 4 to 16 weeks, eventually reaching approximately 2000 mg/dl, a 6- to 7-fold increase from their chow-fed counterparts. Surprisingly, the extent of atherosclerosis in male LDLr−/−/apoA-I−/− compared to male LDLr−/− mice was the same despite a 6-fold higher VLDL and LDL concentration in LDLr−/− mice. Therefore, these results support the concept that HDL apoA-I is highly protective against aortic atherosclerosis and demonstrate that in the presence of plasma HDL the concentration of atherogenic particles, such as LDL, must be significantly higher to induce the same extent of atherosclerosis seen in the absence of plasma HDL.
These dramatic differences in steady-state VLDL and LDL concentration in response to diet may be related to differences in plasma apoE levels, which were 3- to 4-fold higher in chow-fed LDLr−/−/apoA-I−/− compared to chow-fed LDLr−/− mice. The basis for this large difference in plasma apoE has yet to be determined, but appears to be unrelated to the absence of the LDL receptor, since higher plasma apoE concentrations have been reported in apoA-I knockout (KO) mice. 11
In the absence of LDL receptors, receptor-mediated lipoprotein clearance occurs mainly by the liver LDL receptor-related protein, 39 and given the greater concentration of apoE in the LDLr−/−/apoA-I−/− mice, it is possible that LDLr−/−/apoA-I−/− mice catabolize VLDL and LDL more efficiently, resulting in less accumulation of these particles in plasma. However, the liver cholesterol mass was about 5-fold higher in LDLr−/− mice than in LDLr−/−/apoA-I−/− mice. Corroborating the chemical finding, liver sections (Figure 7) ▶ from diet-fed LDLr−/− mice showed numerous large-diameter lipid droplets demonstrating a substantial amount of cholesterol deposition. Therefore, liver cholesterol measurements are consistent with the idea that LDLr−/− mice produce more VLDL and LDL than LDLr−/−/apoA-I−/− mice, which would not be true if higher concentrations of apoE in LDLr−/−/apoA-I−/− mice were responsible for a greater delivery of cholesterol to the liver.
Since plasma apoE levels are higher in chow-fed LDLr−/−/apoA-I−/− mice, it is possible that their lower diet-induced TPC may be a direct result of the absence of plasma apoA-I. In apoA-I KO mice plasma apoE levels are significantly increased compared to control mice suggesting that the absence of apoA-I may alter the production, metabolism, or secretion of apoE. Certain reports suggest that in states of plasma apoE overexpression, hypertriglyceridemia occurs 41 characterized by the overproduction of triglyceride-rich particles from the liver and inhibition of their catabolism. It is also likely that in the absence of apoA-I, less cholesterol is absorbed via the intestine and thus less cholesterol may be available for storage. However, even though LDLr−/−/apoA-I−/− mice had lower TPC and liver cholesterol levels, their skin cholesterol levels were 12-fold higher than LDLr−/− mice, suggesting that dietary cholesterol was absorbed but transported and stored differently in the double-knockout mice.
Typically, female LDLr−/− mice show greater aortic atherosclerosis development than their male counterparts, 40 as was also observed in our study (Table 2) ▶ . However, this gender preference in aortic deposition was not observed when diet-fed female and male LDLr−/−/apoA-I−/− mice were compared. However, our atherosclerosis findings in female LDLr−/−/apoA-I−/− mice may have been influenced by the development of severe inflammation and loss of several mice before the end of the study.
The cause of the fatal inflammation appears to be related to the extensive deposits of cholesterol in the skin of LDLr−/−/apoA-I−/− mice, which showed a 12-fold greater mass of total cholesterol deposited in the skin of both male and female LDLr−/−/apoA-I−/− mice. The most striking increase in skin cholesterol compared to LDLr−/− mice was in the form of cholesterol ester, since the cholesterol found in the skin of diet-fed LDLr−/− mice was predominately in the form of free cholesterol. This fact, combined with a nearly complete depletion of adrenal cholesterol in LDLr−/−/apoA-I−/− mice may form a partial explanation for the onset of this fatal condition.
Microscopically cholesterol deposition in both liver and adrenals of LDLr−/− mice was greater than for LDLr−/−/apoA-I−/− mice, which was verified by tissue mass determination. Microscopic examination of LDLr−/− mouse liver showed numerous large-diameter lipid deposits not observed in LDLr−/−/apoA-I−/− mouse liver. In sharp contrast, LDLr−/−/apoA-I−/− mouse livers showed smaller lipid droplets in the liver, and an infiltration of neutrophils and leukocytes, not seen in either male or female LDLr−/− mice fed the atherogenic diet for 16 weeks. The contribution of the inflammatory reaction to the development of atherosclerosis in the LDLr−/−/apoA-I−/− mouse model is currently being tested.
In conclusion, the current study reveals that LDLr−/−/apoA-I−/− mice do develop severe aortic atherosclerosis in response to an atherogenic diet. This is characterized by a significant increase in plasma apoE, large accumulations of cholesterol ester in the skin, and severe inflammation leading to the accumulation of neutrophils and leukocytes in the liver. Studies are currently underway to determine the basis for the induction of inflammation in this new model of hypercholesterolemia.
Acknowledgments
We thank Janet Sawyer, Persida Tahiri, and Dr. Martha Wilson for expert technical assistance. We also thank Dr. Kim R. Geisinger for counsel regarding the liver histology.
Footnotes
Address reprint requests to Mary G. Sorci-Thomas, Ph.D., Pathology, Wake Forest University Health Sciences, Medical Center Boulevard, Winston-Salem, NC 27157. E-mail: msthomas@wfubmc.edu.
Supported by National Institutes of Health grants HL49373, HL64163, HL64963 (to M.S.T.), and HL-60079 (to M.J.T.).
M.Z. and S.B. contributed equally to this work.
References
- 1.Boden WE: High-density lipoprotein cholesterol as an independent risk factor in cardiovascular disease: assessing the data from Framingham to the Veterans Affairs high-density lipoprotein intervention trial. Am J Cardiol 2000, 86:19L-22L [DOI] [PubMed] [Google Scholar]
- 2.Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DR, Jr, Bangdiwala S, Tyroler HA: High-density lipoprotein cholesterol and cardiovascular disease: four prospective american studies. Circulation 1989, 79:8-15 [DOI] [PubMed] [Google Scholar]
- 3.Navab M, Van Lenten BJ, Reddy ST, Fogelman AM: High-density lipoprotein and the dynamics of atherosclerotic lesions. Circulation 2001, 104:2386-2387 [PubMed] [Google Scholar]
- 4.Walsh A, Ito Y, Breslow JL: High levels of human apolipoprotein A-I in transgenic mice result in increased plasma levels of small high-density lipoprotein (HDL) particles comparable to human HDL3. J Biol Chem 1989, 264:6488-6494 [PubMed] [Google Scholar]
- 5.Rubin EM, Ishida BY, Clift SM, Krauss RM: Expression of human apolipoprotein A-I in transgenic mice results in reduced plasma levels of murine apolipoprotein A-I and the appearance of two new high-density lipoprotein size subclasses. Proc Natl Acad Sci USA 1991, 88:434-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duverger N, Viglietta C, Berthou L, Emmanuel F, Tailleux A, Parmentier-Nihoul L, Laine B, Fievet C, Castro G, Fruchart JC, Houbebine LM, Denèfle P: Transgenic rabbits expressing human apolipoprotein A-I in the liver. Arterioscler Thromb Vasc Biol 1996, 16:1424-1429 [DOI] [PubMed] [Google Scholar]
- 7.Badimon JJ, Badimon L, Fuster V: Regression of atherosclerotic lesions by high-density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest 1990, 85:1234-1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miyazaki A, Sakuma S, Morikawa W, Takiue T, Miake F, Terano T, Sakai M, Hakamata H, Sakamoto YI, Naito M, Ruan YM, Takahashi K, Ohta T, Horiuchi S: Intravenous injection of rabbit apolipoprotein A-I inhibits the progression of atherosclerosis in cholesterol-fed rabbits. Arterioscler Thromb Vasc Biol 1995, 15:1882-1888 [DOI] [PubMed] [Google Scholar]
- 9.Chiesa G, Monteggia E, Marchesi M, Lorenzon P, Laucello M, Lorusso V, Di Mario C, Karvouni E, Newton RS, Bisgaier CL, Franceschini G, Sirtori CR: Recombinant apolipoprotein A-IMilano infusion into rabbit carotid artery rapidly removes lipid from fatty streaks. Circ Res 2002, 90:974-980 [DOI] [PubMed] [Google Scholar]
- 10.Williamson R, Lee D, Hagaman J, Maeda N: Marked reduction of high-density lipoprotein cholesterol in mice genetically modified to lack apolipoprotein A-I. Proc Natl Acad Sci USA 1992, 89:7134-7138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Reddick RL, Maeda N: Lack of apoA-I is not associated with increased susceptibility to atherosclerosis in mice. Arterioscler Thromb 1993, 13:1814-1821 [DOI] [PubMed] [Google Scholar]
- 12.Hughes SD, Verstuyft J, Rubin EM: HDL deficiency in genetically engineered mice requires elevated LDL to accelerate atherogenesis. Arterioscler Thromb Vasc Biol 1997, 17:1725-1729 [DOI] [PubMed] [Google Scholar]
- 13.Voyiaziakis E, Goldberg IJ, Plump AS, Rubin EM, Breslow JL, Huang LS: ApoA-I deficiency causes both hypertriglyceridemia and increased atherosclerosis in human apoB transgenic mice. J Lipid Res 1998, 39:313-321 [PubMed] [Google Scholar]
- 14.Liu AC, Lawn RM, Verstuyft JG, Rubin EM: Human apolipoprotein A-I prevents atherosclerosis associated with apolipoprotein[a] in transgenic mice. J Lipid Res 1994, 35:2263-2267 [PubMed] [Google Scholar]
- 15.Kawashiri MA, Zhang Y, Pure E, Rader DJ: Combined effects of cholesterol reduction and apolipoprotein A-I expression on atherosclerosis in LDL receptor deficient mice. Atherosclerosis 2002, 165:15-22 [DOI] [PubMed] [Google Scholar]
- 16.De Geest B, Zhao Z, Collen D, Holvoet P: Effects of adenovirus-mediated human apoA-I gene transfer on neointima formation after endothelial denudation in apoE-deficient mice. Circulation 1997, 43:4349-4356 [DOI] [PubMed] [Google Scholar]
- 17.Tangirala RK, Praticó D, FitzGerald GA, Chun S, Tsukamoto K, Maugeais C, Usher DC, Puré E, Rader DJ: Reduction of isoprostanes and regression of advanced atherosclerosis by apolipoprotein. Eur J Biol Chem 2001, 276:261-266 [DOI] [PubMed] [Google Scholar]
- 18.Rong JX, Li J, Reis ED, Choudhury RP, Dansky HM, Elmalem VI, Fallon JT, Breslow JL, Fisher EA: Elevating high-density lipoprotein cholesterol in apolipoprotein E-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content. Circulation 2001, 104:2447-2452 [DOI] [PubMed] [Google Scholar]
- 19.Benoit P, Emmanuel F, Caillaud JM, Bassinet L, Castro G, Gallix P, Fruchart JC, Branellec D, Denèfle P, Duverger N: Somatic gene transfer of human apoA-I inhibits atherosclerosis progression in mouse models. Circulation 1999, 99:105-110 [DOI] [PubMed] [Google Scholar]
- 20.Plump AS, Scott CJ, Breslow JL: Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci USA 1994, 91:9607-9611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pászty C, Maeda N, Verstuyft J, Rubin EM: Apolipoprotein AI transgene corrects apolipoprotein E deficiency-induced atherosclerosis in mice. J Clin Invest 1994, 94:899-903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sorci-Thomas MG, Thomas MJ: The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med 2002, 12:121-128 [DOI] [PubMed] [Google Scholar]
- 23.Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J: Hypercholesterolemia in low-density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest 1993, 92:883-893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK: Massive xanthomatosis and atherosclerosis in cholesterol-fed low-density lipoprotein receptor-negative mice. J Clin Invest 1994, 93:1885-1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudel LL, Kelley K, Sawyer JK, Shah R, Wilson MD: Dietary monounsaturated fatty acids promote aortic atherosclerosis in LDL receptor - null, human apoB100 - overexpression transgenic mice. Arterioscler Thromb Vasc Biol 1998, 18:1818-1827 [DOI] [PubMed] [Google Scholar]
- 26.Furbee JW, Sawyer JK, Parks JS: Lecithin: cholesterol acyltransferase (LCAT) deficiency increases atherosclerosis in the low-density lipoprotein receptor (LDLr) or apolipoprotein E (apoE) knockout mice. J Biol Chem 2002, 277:3511-3519 [DOI] [PubMed] [Google Scholar]
- 27.Warnick GR: Dextran sulfate-Mg2+ precipitation procedure for quantification of high-density lipoprotein cholesterol. Clin Chem 1982, 28:1379-1388 [PubMed] [Google Scholar]
- 28.Sorci-Thomas MG, Thomas M, Curtiss L, Landrum M: Single repeat deletion in apoA-I blocks cholesterol esterification and results in rapid catabolism of Δ6 and wild-type apoA-I in transgenic mice. J Biol Chem 2000, 275:12156-12163 [DOI] [PubMed] [Google Scholar]
- 29.Lowry OJ, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 1951, 19:265-275 [PubMed] [Google Scholar]
- 30.Parks JS, Li H, Gebre AK, Smith TL, Maeda N: Effect of apolipoprotein A-I deficiency on lecithin: cholesterol acyltransferase activation in mouse plasma. J Lipid Res 1995, 36:349-355 [PubMed] [Google Scholar]
- 31.Ishibashi S, Herz J, Maeda N, Goldstein JL, Brown MS: The two-receptor model of lipoprotein clearance: tests of the hypothesis in “knockout” mice lacking the low-density lipoprotein receptor, apolipoprotein E, or both proteins. Proc Natl Acad Sci USA 1994, 91:4431-4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Véniant MM, Sullivan MA, Kim SK, Ambroziak P, Chu A, Wilson MD, Hellerstein MK, Rudel LL, Walzem RL, Young SG: Defining the atherogenicity of large and small lipoproteins containing apolipoprotein B100. J Clin Invest 2000, 106:1501-1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Accad M, Smith SJ, Newland DL, Sanan DA, King LE, Jr, Linton MF, Fazio S, Farese RV, Jr: Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA: cholesterol acyltransferase 1. J Clin Invest 2000, 105:711-719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yagyu H, Kitamine T, Osuga J, Tozawa R, Chen Z, Kaji Y, Oka T, Perrey S, Tamura Y, Ohashi K, Okaxaki H, Yahagi N, Shionoiri F, Iizuka Y, Harada K, Shimano H, Yamashita H, Gotoda T, Yamada N, Ishibashi S: Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J Biol Chem 2000, 275:21324-21330 [DOI] [PubMed] [Google Scholar]
- 35.Aiello RJ, Brees D, Bourassa PA, Royer L, Lindsey S, Coskran T, Haghpassand M, Francone OL: Increased atherosclerosis in hyperlipidemic mice with inactivation of ABCA1 in macrophages. Arterioscler Thromb Vasc Biol 2002, 22:630-637 [DOI] [PubMed] [Google Scholar]
- 36.Plump AS, Erickson SK, Weng W, Partin JS, Breslow JL, Williams DL: Apolipoprotein A-I is required for cholesteryl ester accumulation in steroidogenic cells and for normal adrenal steroid production. J Clin Invest 1996, 97:2660-2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gwynne JT, Hess B: The role of high-density lipoproteins in rat adrenal cholesterol metabolism and steroidogenesis. J Biol Chem 1980, 255:10875-10883 [PubMed] [Google Scholar]
- 38.Gynne JT, Mahaffee DD: Rat adrenal uptake and metabolism of high-density lipoproteins cholesteryl ester. J Biol Chem 1989, 264:8141-8150 [PubMed] [Google Scholar]
- 39.Willnow TE, Armstrong SA, Hammer RE, Herz J: Functional expression of low-density lipoprotein receptor-related protein is controlled by receptor-associated protein in vivo. Proc Natl Acad Sci USA 1995, 92:4537-4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merkel M, Velez-Carrasco W, Hudgins LC, Breslow JL: Compared with saturated fatty acids, dietary monounsaturated fatty acids and carbohydrates increase atherosclerosis and VLDL cholesterol levels in LDL receptor-deficient, but not apolipoprotein E-deficient, mice. Proc Natl Acad Sci USA 2001, 98:13294-13299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Y, Liu XQ, Rall SC, Taylor JM, von Eckardstein A, Assman G, Mahley RW: Overexpression and accumulation of apoE as a cause of hypertriglyceridemia. J Biol Chem 1998, 273:26388-26393 [DOI] [PubMed] [Google Scholar]