

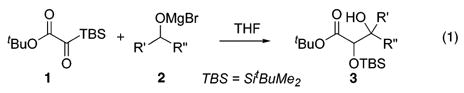
The aldol reaction is the preeminent method for the introduction of the β-hydroxy carbonyl function, and its development has been marked by significant advances in utility to synthetic practitioners.1 The most recent chapter in this evolution is the introduction of catalysts and reagents that enable direct and selective formation of the nucleophilic enol component in the presence of the carbonyl electrophile.2 Although electrophile synthesis is not typically factored into the overall efficiency of a given aldol addition, it is instructive to consider that when the reaction is applied in complex fragment couplings, the aldolization step is often preceded by an obligatory oxidation event that provides the requisite aldehyde or ketone coupling partner.3 A compelling argument may be advanced, therefore, that the most efficient direct aldol reaction would be one in which both the enolate nucleophile and carbonyl electrophile are simultaneously generated in situ. This communication provides the conceptual framework for such a process in the form of a symbiotic redox reaction between an alcohol and a silylglyoxylate that mutually activates both reaction components for aldolization in the second stage (eq 1).
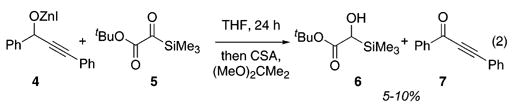
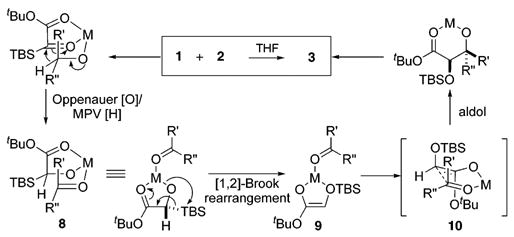
Silylglyoxylates4 1 and 5 were recently described as useful conjunctive agents for coupling alkynylzinc halides and aldehydes.5 The genesis of the current study was the observation of hydroxy-silane 6 and ynone 7 as minor byproducts in a reaction between zinc alkoxide 4 and silyl glyoxylate 5 (eq 2) that was designed to probe the mechanism of the aforementioned three-component coupling. We hypothesized that these byproducts resulted from an Oppenauer oxidation/Meerwein–Ponndorf–Verley (MPV) reduction6–8 between 4 and 5. In contrast to other nucleophiles that react with 5, the hydride transfer did not trigger [1,2]-Brook rearrangement.9
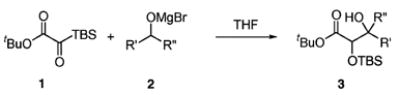
If reaction conditions could be suitably modified such that the Oppenauer/MPV process did cause C → O silyl migration (8 → 9),10 the resulting products from the redox reaction would be a glycolate enolate and ketone or aldehyde poised to undergo aldolization (Scheme 1). It was projected that the identity of the metal cation would be crucial in governing the efficiency of each proposed step; therefore, an evaluation of suitable candidates was initiated.
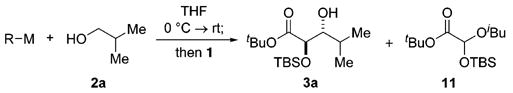
As commonly employed catalysts for MPV/Oppenauer reactions, aluminum alkoxides provided a logical starting point for this inquiry (Table 1).11 Surprisingly, we observed no reaction with MeAlCl2 (entry 1), while nBuLi and Bu3La provided only the direct addition/rearrangement product 11 (entries 2 and 3). Selective generation of desired aldol product 3a was achieved with a magnesium alkoxide12,13 generated in THF (entry 4), and an improved yield and diastereomer ratio was realized when the reaction was conducted in 2:1 THF/CH2Cl2 (entry 5).14
Table 1.
Evaluation of Metal Alkoxides
| entry | R–M | result | anti:syna |
|---|---|---|---|
| 1 | MeAlCl2 | no reaction | n.a. |
| 2 | n-BuLi | 40% of 11b | n.a. |
| 3 | Bu3La | 58% of 11c | n.a. |
| 4 | EtMgBr | 71% of 3ac | 6:1 |
| 5 | EtMgBr | 97% of 3ac,d | 10:1 |
Determined by 1H NMR spectroscopy.
1H NMR yield versus an internal standard.
Isolated yield.
Reaction solvent: 2:1 THF/CH2Cl2.
With the identification of the optimal metal cation, we next evaluated other coupling partners in the reaction. Alkoxides resulting from deprotonation of alcohols with EtMgBr were initially investigated (Table 2). Results were good for a variety of alcohols with yields from 63 to 97%. Notably, primary aliphatic alcohols delivered the aldol products with synthetically useful levels of anti diastereocontrol (Table 2, entries 1–4).15 Although the details of the transition structure will require further elucidation, the predominance of the anti isomer is congruent with the recent observation by Evans and co-workers of anti propionates from (Z)-magnesium enolates.16 The boat-like transition structure 10 may thus be construed as a tentative model for the observed stereochemical outcome (R′ = H).
Table 2.
Oppenauer Oxidation/Brook Rearrangement/Aldolization Reactionsa
| entry | alcohol | product | yield (%)b | d.r.c |
|---|---|---|---|---|
| 1d | Me2CHCH2OH |
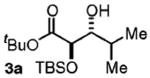
|
97 | 10:1 |
| 2d | Me(CH2)5OH |
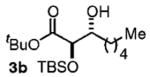
|
86 | 7:1 |
| 3d | TMS(CH2)3OH |
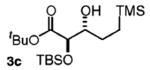
|
88 | 5:1 |
| 4 | CH2=CH(CH2)4OH |
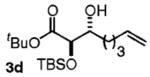
|
63 | 5:1 |
| 5 | PhCH2OH |
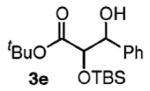
|
90 | 1.2:1 |
| 6 | 4-ClPhCH2OH |
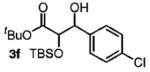
|
82 | 1:1 |
| 7 | 4-MeOPhCH2OH |
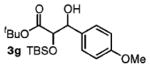
|
85 | 1:1 |
| 8 | PhCH(OH)Me |
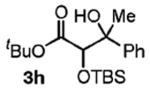
|
67 | 2.5:1 |
| 9 | cyclohexanon |
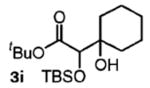
|
68 | n.a. |
Alcohol (1.5 equiv), EtMgBr (2.0 equiv), 0 °C → rt; then 1 (1.0 equiv).
Isolated yield.
Determined by 1H NMR spectroscopy; the major isomer is shown.
Reaction solvent: 2:1 THF/CH2Cl2.
Benzylic alcohols provided the aldol adducts with superior yields but negligible diastereocontrol (entries 5–7). Perhaps most strikingly, secondary alcohols function effectively in this reaction to deliver highly substituted ketone aldol adducts (entries 8 and 9).
The success of these latter reactions led us to evaluate a three-component coupling strategy wherein the requisite secondary alkoxide was formed via Grignard addition to aldehydes (Table 3).17 This simple one-step protocol facilitated access to more complex ketone aldol adducts with no reduction in reaction efficiency. In the case where significant steric differentiation exists between R′ and R″, promising levels of diastereocontrol may be achieved (entry 3).
Table 3.
Reaction Initiation via Aldehyde Alkylation
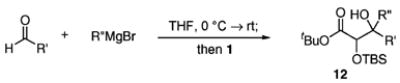
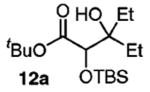
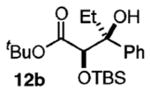
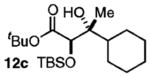
Isolated yields.
Determined by 1H NMR spectroscopy; the major isomer is shown.
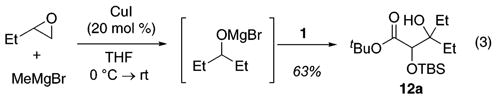
Epoxides may also serve as the alkoxide progenitor in conjunction with a Cu(I)-catalyzed alkylation (eq 3). On the basis of the similar yield for 12a beginning from either an epoxide (eq 3) or an aldehyde (Table 3, entry 1), it appears that CuI does not interfere with the subsequent steps.
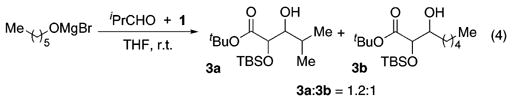
Preliminary conclusions regarding the relative rates of the individual steps of the reaction sequence may be drawn from a simple crossover experiment shown in eq 4. Exposing the magnesium alkoxide of n-hexanol to 1 and isobutyraldehyde resulted in an approximately equimolar mixture of aldols 3a and 3b, revealing that dissociation of the aldehyde from the magnesium center is faster than Brook rearrangement and aldolization (eq 4).18
In summary, a new direct aldol reaction has been accomplished between the enolate obtained from an Oppenauer/MPV-induced [1,2]-Brook rearrangement of a silylglyoxylate and the carbonyl product of that redox reaction. The concept of symbiotic reagent activation may be applicable to other reaction classes. This possibility is the topic of ongoing investigations.
Supplementary Material
Scheme 1.
Acknowledgments
Funding for this work was provided by the National Institutes of Health (National Institute of General Medical Sciences, GM068443). Research support from Eli Lilly, Amgen, and GSK is gratefully acknowledged. J.S.J. is an Alfred P. Sloan Fellow and a Camille Dreyfus Teacher-Scholar.
Footnotes
Supporting Information Available: Experimental procedures and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mahrwald R, editor. Modern Aldol Reactions. Wiley-VCH; Weinheim, Germany: 2004. [Google Scholar]
- 2.For leading references, see: Yoshikawa N, Yamada YMA, Das J, Sasai H, Shibasaki M. J Am Chem Soc. 1999;121:4168–4178.Taylor SJ, Morken JP. J Am Chem Soc. 1999;121:12202–12203.List B, Lerner RA, Barbas CF., III J Am Chem Soc. 2000;122:2395–2396.Trost BM, Ito H. J Am Chem Soc. 2000;122:12003–12004.Northrup AB, MacMillan DWC. J Am Chem Soc. 2002;124:6798–6799. doi: 10.1021/ja0262378.Jang HY, Huddleston RR, Krische MJ. J Am Chem Soc. 2002;124:15156–15157. doi: 10.1021/ja021163l.Evans DA, Downey CW, Hubbs JL. J Am Chem Soc. 2003;125:8706–8707. doi: 10.1021/ja035509j.Ooi T, Kameda M, Taniguchi M, Maruoka K. J Am Chem Soc. 2004;126:9685–9694. doi: 10.1021/ja048865q.
- 3.For a recent example, see: Kagawa N, Ihara M, Toyota M. Org Lett. 2006;8:875–878. doi: 10.1021/ol052943c.
- 4.Bolm C, Kasyan A, Heider P, Saladin S, Drauz K, Guenther K, Wagner C. Org Lett. 2002;4:2265–2267. doi: 10.1021/ol025911n. [DOI] [PubMed] [Google Scholar]
- 5.Nicewicz DA, Johnson JS. J Am Chem Soc. 2005;127:6170–6171. doi: 10.1021/ja043884l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Graauw CF, Peters JA, van Bekkum H, Huskens J. Synthesis. 1994:1007–1017. [Google Scholar]
- 7.Adkins H, Franklin RC. J Am Chem Soc. 1941;63:2381–2383. [Google Scholar]
- 8.Markert M, Mahrwald R. Synthesis. 2004:1429–1433. [Google Scholar]
- 9.For reduction of acyl silanes by organometals and metal hydrides, see: Brook AG, Quigley MA, Peddle GJD, Schwartz NV, Warner CM. J Am Chem Soc. 1960;82:5102–5106.Reich HJ, Holtan RC, Bolm C. J Am Chem Soc. 1990;112:5609–5617.
- 10.Brook AG. Acc Chem Res. 1974;7:77–84. [Google Scholar]
- 11.Ooi T, Otsuka H, Miura T, Ichikawa H, Maruoka K. Org Lett. 2002;4:2669–2672. doi: 10.1021/ol020094c. [DOI] [PubMed] [Google Scholar]
- 12.Oppenauer R. US Patent 2,384,335. 1945. [Google Scholar]
- 13.The absence of silyl migration in eq 2 and the successful use of a magnesium alkoxide to promote [1,2]-Brook rearrangement (Table 1, entry 4) contrast a recent example where Mg → Zn transmetalation was required to initiate silyl migration: Unger R, Cohen T, Marek I. Org Lett. 2005;7:5313–5316. doi: 10.1021/ol052237b.
- 14.For a complete survey of conditions, see the Supporting Information.
- 15.(a) Notz W, List B. J Am Chem Soc. 2000;122:7386–7387. [Google Scholar]; (b) Andrus MB, Sekhar BBVS, Meredith EL, Dalley NK. Org Lett. 2000;2:3035–3037. doi: 10.1021/ol0002166. [DOI] [PubMed] [Google Scholar]; (c) Crimmins MT, McDougall PJ. Org Lett. 2003;5:591–594. doi: 10.1021/ol034001i. [DOI] [PubMed] [Google Scholar]; (d) Northrup AB, Mangion IK, Hettche F, MacMillan DWC. Angew Chem, Int Ed. 2004;43:2152–2154. doi: 10.1002/anie.200453716. [DOI] [PubMed] [Google Scholar]
- 16.Evans DA, Downey CW, Shaw JT, Tedrow JS. Org Lett. 2002;4:1127–1130. doi: 10.1021/ol025553o. [DOI] [PubMed] [Google Scholar]
- 17.Byrne B, Karras M. Tetrahedron Lett. 1987;28:769–772. [Google Scholar]
- 18.A separate control experiment between nhexOMgBr and iPrCHO yielded <3% (GLC) of nhexanal, indicating that the mechanism of crossover is not MPV/Oppenauer redox between the alkoxide and the aldehyde.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.