
Figure 1.
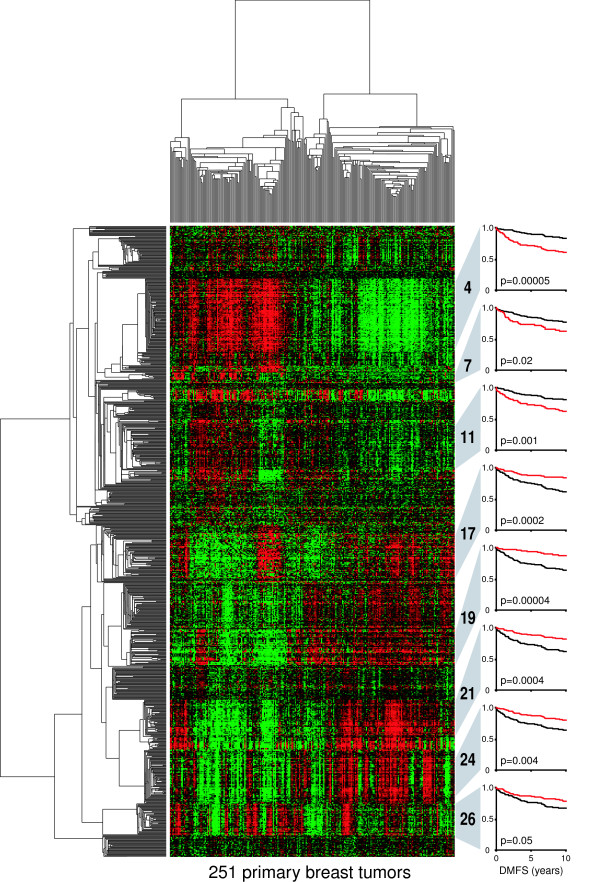
Clustergram of diverse gene expression signatures prognostic of breast cancer recurrence. Tumors (n = 251; columns) and gene probe sets (n = 816; rows) of the Uppsala cohort (GEO ID GSE3494) [26] were hierarchically clustered by using Pearson correlation and average linkage analysis. Probe-set values were natural-log-transformed and mean centered before clustering. Initially, all 44,928 probe sets (on the Affymetrix U133A and U133B arrays) were assessed for survival correlations as follows. The expression value for each gene was used to dichotomize patients into below-mean and above-mean expression groups. The two groups were then assessed for differences in distant metastasis-free survival (DMFS) by Cox regression analysis. Probe sets significantly associated with DMFS (that is, with likelihood-ratio test P values of less than 0.05) were hierarchically clustered as described above, and clusters with average correlations of more than 0.5 were selected for inclusion in the figure. Probe sets within clusters were then averaged for each tumor, and cluster survival associations were determined as described above. Kaplan-Meier plots for selected numbered clusters are shown at the right. The red survival curves indicate patients with above-mean cluster expression. Cluster 7 is composed of five genes, all mapping to chromosome 17q12 (ORMDL3, PSMD3, CRKRS, PERLD1, and C17ORF37) with an average expression correlation of 0.64. Cluster 11, with an average correlation of 0.65, consists of 31 distinct genes, 18 of which map to chromosome 16p13 (PPP4C, PARN, ATP6V0C, C16orf14, GBL, HAGH, ITFG3, MGC13114, MRPS34, NDUFB10, NMRAL1, NTHL1, NUBP2, POLR3K, RNPS1, STUB1, TBL3, and USP7).