

Abstract
Aims
The aim of the study was to clarify whether the pharmacokinetic interaction between theophylline and mexiletine is mediated by inhibition of CYP1A2 and to assess the possible interaction potential of other antiarrhythmic drugs with drugs metabolized by CYP1A2.
Methods
The inhibitory effects of mexiletine and 10 antiarrhythmic drugs on phenacetin O-deethylation, a marker reaction of CYP1A2, were studied using human liver microsomes and cDNA-expressed CYP1A2.
Results
Propafenone and mexiletine inhibited phenacetin O-deethylation with IC50 values of 29 and 37 μm, respectively. Disopyramide, procainamide and pilsicainide produced negligible inhibition of phenacetin O-deethylation (IC50>1 mm). Amiodarone, bepridil, aprindine, lignocaine, flecainide and quinidine inhibited phenacetin O-deethylation in a concentration-dependent manner, although the inhibitory effects were relatively weak with IC50 values ranging from 86 to 704 μm. Propafenone and mexiletine selectively abolished the high-affinity component of phenacetin O-deethylation in human liver microsomes. In addition, propafenone and mexiletine inhibited phenacetin O-deethylation catalysed by cDNA-expressed CYP1A2.
Conclusions
These data suggest that, among the antiarrhythmic drugs studied, propafenone and mexiletine are relatively potent inhibitors of CYP1A2, which may cause a drug-drug interaction with drugs metabolized by CYP1A2.
Keywords: CYP1A2, phenacetin O-deethylation, human liver microsomes, mexiletine, propafenone
Introduction
Cytochrome P450 1A2 (CYP1A2), an isoform of the CYP1A subfamily [1], accounts for about 10 to 15% of the total CYP content of human livers [2]. This enzyme is known to catalyse the metabolism of imipramine [3], propranolol [4], clozapine [5], caffeine [6], theophylline [7] and phenacetin [6, 8]. The kinetic disposition of these drugs is altered by the administration or exposure to inducers or inhibitors of CYP1A2. For example, the clearance of propranolol [9], clozapine [10] and theophylline [11] is greater in smokers than in nonsmokers, as a result of marked induction of CYP1A2 by cigarette smoking. On the other hand, the plasma concentrations of imipramine [12], clozapine [13], caffeine [14, 15] and theophylline [16] are increased after the co-administration of drugs such as furafylline [17] and fluvoxamine [18], which inhibit CYP1A2 [17, 18].
Theophylline is occasionally coadministered with antiarrhythmic drugs in patients with cardiac arrhythmias and chronic or obstructive pulmonary disease. Mexiletine, a class I antiarrhythmic drug, has been reported to elevate plasma levels of concomitantly administered theophylline [19–24], by inhibiting 1- and 3-demethylation and 8-hydroxylation of theophylline [19, 20]. Recent studies using human liver microsomes and cDNA-expressed human CYP isoforms indicated that CYP1A2 is the enzyme mainly responsible for the 1- and 3-demethylation and 8-hydroxylation of theophylline in humans [25, 26]. Several studies using rat liver microsomes also showed that mexiletine inhibits ethoxyresorufin O-dealkylation catalysed by CYP1A1, another isoform of the CYP1A subfamily [27]. However, in vivo [28, 29] and in vitro [30, 31] studies showed that mexiletine is mainly metabolized by CYP2D6. Therefore, it remains obscure as to whether the pharmacokinetic interaction between theophylline and mexiletine occurs by competitive inhibition of CYP1A2 in humans.
In the present study, our objective was to study the inhibitory effects of mexiletine on phenacetin O-deethylation, a marker reaction of CYP1A2, in human liver microsomes and cDNA-expressed CYP1A2. We also studied the inhibitory effects of 10 other antiarrhythmic drugs on phenacetin O-deethylation in human liver microsomes in order to assess their possible interaction potential with drugs metabolized via CYP1A2 (e.g., theophylline) when administered concurrently.
Methods
Drugs
Mexiletine hydrochloride was kindly supplied by Nihon Boehringer Ingelheim Co. Ltd (Hyogo, Japan), aprindine hydrochloride by Mitsui Pharmaceutical Industries Ltd (Tokyo, Japan), flecainide acetate by Eisai Co. Ltd (Tokyo, Japan), amiodarone hydrochloride by Taisho Pharmaceutical Co. Ltd (Tokyo, Japan) and pilsicainide hydrochloride by Suntory Co. Ltd (Osaka, Japan). Bepridil hydrochloride, procainamide hydrochloride and disopyramide phosphate were purchased from Research Biochemicals International (Natick, MA, USA). Propafenone hydrochloride was purchased from Sigma Chemical Company (St Louis, MO, USA). NADP+ and glucose-6-phosphate were purchased from Oriental Yeast Co. Ltd (Tokyo, Japan). Glucose-6-phosphate dehydrogenase was obtained from Boehringer Mannheim GmbH (Mannheim, Germany). Phenacetin, paracetamol (acetaminophen), caffeine, lignocaine hydrochloride, quinidine, acetonitrile and other reagents of analytical grade were purchased from Wako Pure Chemical Industries Ltd (Osaka, Japan).
Microsomes
Three human liver samples (HL-32, 33 and 34) were obtained from Japanese patients who underwent partial hepatectomy at the Division of General Surgery, Department of Surgery, International Medical Center of Japan (Tokyo, Japan) as reported from our laboratory [32, 33]. All surgical procedures were performed for the removal of metastatic tumor(s) from the liver. The use of human liver tissue for this study was approved by the Institutional Ethics Committee of the International Medical Center of Japan (Tokyo, Japan). The mean (±s.d.) activity of phenacetin O-deethylation in the three human liver microsomal samples assessed by the method described below was 0.184±0.070 nmol mg−1 min−1, when 10 μm phenacetin was used as a substrate concentration.
Microsomes from human B-lymphoblastoid cells expressing human CYP1A2 were purchased from Gentest Corp. (Woburn, MA, USA). The CYP1A2 content of microsomes was 128 pmol P450 mg −1 protein.
Phenacetin O-deethylation activity
The microsomal phenacetin O-deethylation activity was determined by measuring the rate of paracetamol formation at 37° C by a modification of a high-performance liquid chromatography (h.p.l.c.) method reported previously [32]. Briefly, the h.p.l.c. system consisted of a model L-7100 pump (Hitachi Ltd, Tokyo, Japan), a model L-7420 u.v. detector (Hitachi), a model L-7200 autosampler (Hitachi), a model D-7500 integrator (Hitachi) and a 4.6×250 mm CAPCELL PAK C18 UG120 column (Shiseido Co. Ltd, Tokyo, Japan). The mobile phase consisted of 0.05 m potassium dihydrogenphosphate and acetonitrile at a ratio of 85/15 by v/v (%) and was delivered at a flow rate of 0.7 ml min−1. The eluate was monitored at a wavelength of 245 nm. The column temperature was maintained at 30° C. Calibration curves were generated from 31.3 to 312.5 pmol ml−1 by processing the authentic standard substances through the entire procedures. The linearity of calibration curve was confirmed to be retained down to 6 pmol ml−1. Analyte was quantified by comparison with the standard curves using the peak-height ratio method.
Incubation medium contained 0.025 mg protein of human liver microsomes, 100 mm potassium phosphate buffer (pH 7.4), 0.1 mm EDTA, NADPH generating system (0.5 mm NADP+, 2.0 mm glucose-6-phosphate, 1 IU ml−1 of glucose-6-phosphate dehydrogenase, 4 mm MgCl2), and phenacetin in the presence or absence of one of antiarrhythmic drugs in a final volume of 250 μl. Phenacetin and antiarrhythmic drugs dissolved in methanol were added to test tubes and evaporated with vacuum evaporator at 40° C. The incubation mixture, excepting for microsomes and the NADPH generating system, were added and the compounds were redissolved by sonication. The mixture including microsomes and NADPH generating system was incubated at 37°C for 30 min, because the formation of paracetamol under the condition as described above was linear for up to 60 min. After the reaction was stopped by adding 100 μl of cold acetonitrile, 50 μl of caffeine (5 μg ml−1 in methanol) was added to the samples as an internal standard. The mixture was centrifuged at 10 000 g for 5 min, and the supernatant was evaporated by vacuum evaporator at 40° C for 15 min. Fifty μl of the remaining sample was injected into the h.p.l.c. system as described above.
Effects of antiarrhythmic drugs on phenacetin O-deethylation in human liver microsomes
The effects of antiarrhythmic drugs (i.e., mexiletine, propafenone, bepridil, aprindine, lignocaine, flecainide, quinidine, amiodarone, pilsicainide, procainamide and disopyramide) on phenacetin (10 μm) O-deethylation were studied in microsomes from three human livers. In this part of the study, the concentration of antiarrhythmic drugs used was 30 μm. The concentration-inhibition relationship of each antiarrhythmic drug on phenacetin O-deethylation activity was studied using human liver microsomes, HL-33. The concentrations of antiarrhythmic drugs used were 0, 10, 30, 100, 300 and 1000 μm.
Kinetic analysis of phenacetin O-deethylation in human liver microsomes
Nine different concentrations of phenacetin, ranging from 1 to 200 μm, were used in the kinetic experiments for phenacetin O-deethylation by microsomes from three human livers. Among the eleven antiarrhythmic drugs studied, mexiletine and propafenone more strongly inhibited phenacetin O-deethylation in human liver microsomes compared with the remaining nine drugs. Therefore, the enzyme kinetics for phenacetin O-deethylation were compared with those in the presence of 30 μm mexiletine or propafenone.
Michaelis-Menten parameters for phenacetin O-deethylation were estimated by a two-enzyme kinetic approach. The parameters were calculated initially by graphic analysis of Eadie-Hofstee plots, and the values obtained were used as the first estimate for the nonlinear least-squares regression analysis, MULTI [33], in which unweighted raw data were fitted to the following model equation:
where V is the velocity of phenacetin O-deethylation, S is the concentration of phenacetin, Km1 and Km2 are the affinity constants for the high- and low-affinity components and Vmax1 and Vmax2 are the maximum enzyme velocities for the high- and low-affinity components, respectively. In the case that Eadie-Hofstee plots showed a simple Michaelis-Menten kinetic behaviour, the kinetic parameters (Km, Vmax and Vmax/Km without the subindices) were estimated by linear regression analysis using unweighted raw data.
Inhibition of mexiletine and propafenone on phenacetin
O-deethylation in recombinant CYP1A2
Inhibition of phenacetin O-deethylation by mexiletine or propafenone was also studied in microsomes from human B-lymphoblastoid cells expressing human CYP1A2. The formation rate of paracetamol from phenacetin by recombinant CYP1A2 was linear for up to 20 min. Accordingly, microsomes expressing CYP1A2 (0.05 mg protein) were incubated at 37° C for 20 min with phenacetin (5, 10, 25 and 50 μm) in the presence of mexiletine (0, 10, 25 and 50 μm) or propafenone (0, 10, 25 and 50 μm), respectively. Inhibition patterns were determined by a visual inspection of double reciprocal plots for phenacetin concentration vs the velocity of the reaction. Apparent Km in each concentration of the putative inhibitors was estimated by linear regression analysis using unweighted raw data following a simple (one-enzyme) Michaelis-Menten kinetic approach. The Ki (inhibition constant) values were determined from unweighted linear regression analysis using the following equation [34, 35];
where Km is the affinity constant in the absence of an inhibitor, and apparent Km is the Km in the presence of an inhibitor, and I is the inhibitor concentration.
Results
Effects of antiarrhythmic drugs on phenacetin O-deethylation in human liver microsomes
The effects of eleven antiarrhythmic drugs on the O-deethylation activities of 10 μm phenacetin in human liver microsomes are shown in Figure 1. The mean (±s.d.) inhibition percentage values were 58.5±2.8, 46.2±11.8, 28.6±8.1, 26.6±4.5, 19.9±3.0, 15.1±4.5, 12.5±8.4, 11.6±5.6, 10.8±6.7, 7.2±5.1 and 6.5±6.0% for 30 μm of propafenone, mexiletine, amiodarone, bepridil, aprindine, flecainide, quinidine, disopyramide, pilsicainide, procainamide and lignocaine, respectively. Figure 2 shows the concentration effects of these antiarrhythmic drugs on the O-deethylation activities of 10 μm phenacetin in a human liver microsomal sample (HL-33). The IC50 values obtained are listed in Table 1. Among the antiarrhythmic drugs studied, propafenone and mexiletine more strongly inhibited the phenacetin O-deethylation activities with IC50 values of 29 and 37 μm, respectively (Figure 2a) as compared with other drugs (Figure 2b and 2c). Amiodarone, bepridil, aprindine, lignocaine, flecainide and quinidine inhibited the activities in a concentration-dependent manner, with IC50 values ranging from 86 to 704 μm, indicating that they are weak inhibitors against phenacetin O-deethylation (Figure 2b). Disopyramide, procainamide and pilsicainide scarcely inhibited the activities with IC50 values more than 1000 μm (Figure 2c).
Figure 1.

Effects of eleven antiarrhythmic drugs on phenacetin O-deethylation activity in human liver microsomes. Each bar represents the mean±s.d. data obtained from three different livers (HL-32, 33 and 34) with each liver studied in duplicate microsomal samples.
Figure 2.

Concentration-effect relationship of mexiletine (○) and propafenone (•) (a), amiodarone (•), bepridil (▴), aprindine (▪), lignocaine (○), flecainide (▵) and quinidine (□) (b) and disopyramide (•), procainamide (▴) and pilsicainide (▪) (c) on phenacetin O-deethylation activities in human liver microsomes (HL-33). Each data point represents the average of duplicate determinations.
Table 1.
IC50 values for phenacetin O-deethylation by eleven antiarrhythmic drugs in human liver microsomes (HL-33).

Kinetics of phenacetin O-deethylation in human liver microsomes
Typical Eadie-Hofstee plots for phenacetin O-deethylation in human liver microsomes (HL-34) are shown in Figure 3. The plots observed in the absence of mexiletine or propafenone showed a biphasic curve, suggesting that at least two enzymes were involved in phenacetin O-deethylation (Figure 3a). Eadie-Hofstee plots in two other liver microsomal samples (HL-32 and 33) also showed biphasic curves. Therefore, the kinetic parameters were estimated by assuming that the metabolic reaction was catalysed by the two enzymes. The kinetic parameters calculated by the two-enzyme kinetic approach (Km1, Km2, Vmax1, Vmax2, Vmax1/Km1 and Vmax2/Km2) in the absence of either of the antiarrhythmics are listed in Table 2. The Km values ranged from 1.7 to 4.1 μm and from 23.4 to 65.1 μm for the high- and low-affinity component, respectively. On the other hand, Eadie-Hofstee plots in the presence of 30 μm mexiletine (Figure 3b) or propafenone (Figure 3c) showed a simple Michaelis-Menten kinetic behaviour. The parameters (Km, Vmax and Vmax/Km) in the presence of mexiletine or propafenone are listed in Tables 3 and 4, respectively. The respective Km value in the presence of mexiletine (Table 3) or propafenone (Table 4) was similar to the low-affinity component Km value (Km2) for phenacetin O-deethylation in the absence of mexiletine or propafenone (Table 2).
Figure 3.

Representative Eadie-Hofstee plots for phenacetin O-deethylation in the absence (a) or presence of mexiletine (b) and propafenone (c) in human liver microsomes (HL-34). The solid line drawn in (a) is the computer-generated curve of best fit for a two-enzyme kinetic model, while the lines in b) and c) drawn indicate the best fit for a one-enzyme kinetic model.
Table 2.
Michaelis-Menten parameters of phenacetin O-deethylation in microsomes from three human livers.
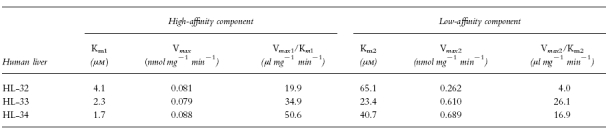
Table 3.
Michaelis-Menten parameters of phenacetin O-deethylation in the presence of 30 μm mexiletine in microsomes from three human livers.

Table 4.
Michaelis-Menten parameters of phenacetin O-deethylation in the presence of 30 μm propafenone in microsomes from three human livers.
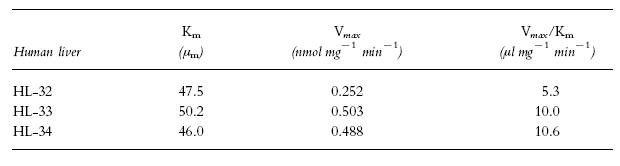
Inhibition of mexiletine and propafenone on phenacetin
O-deethylation in recombinant CYP1A2
In microsomes from B-lymphoblastoid cells expressing human CYP1A2, phenacetin was O-deethylated with Km and Vmax values of 12 μm and 2.8 pmol pmol−1 P450 min−1, respectively. Mexiletine and propafenone inhibited the activities. The inhibition percentage values of phenacetin O-deethylation, when 10 μm phenacetin was used as a substrate, were 35.2 and 53.6% for 50 μm of mexiletine and propafenone, respectively. The inhibition occurred in a competitive manner for both mexiletine and propafenone as shown in Figure 4. The Ki values of mexiletine and propafenone were 47 and 21 μm, respectively.
Figure 4.
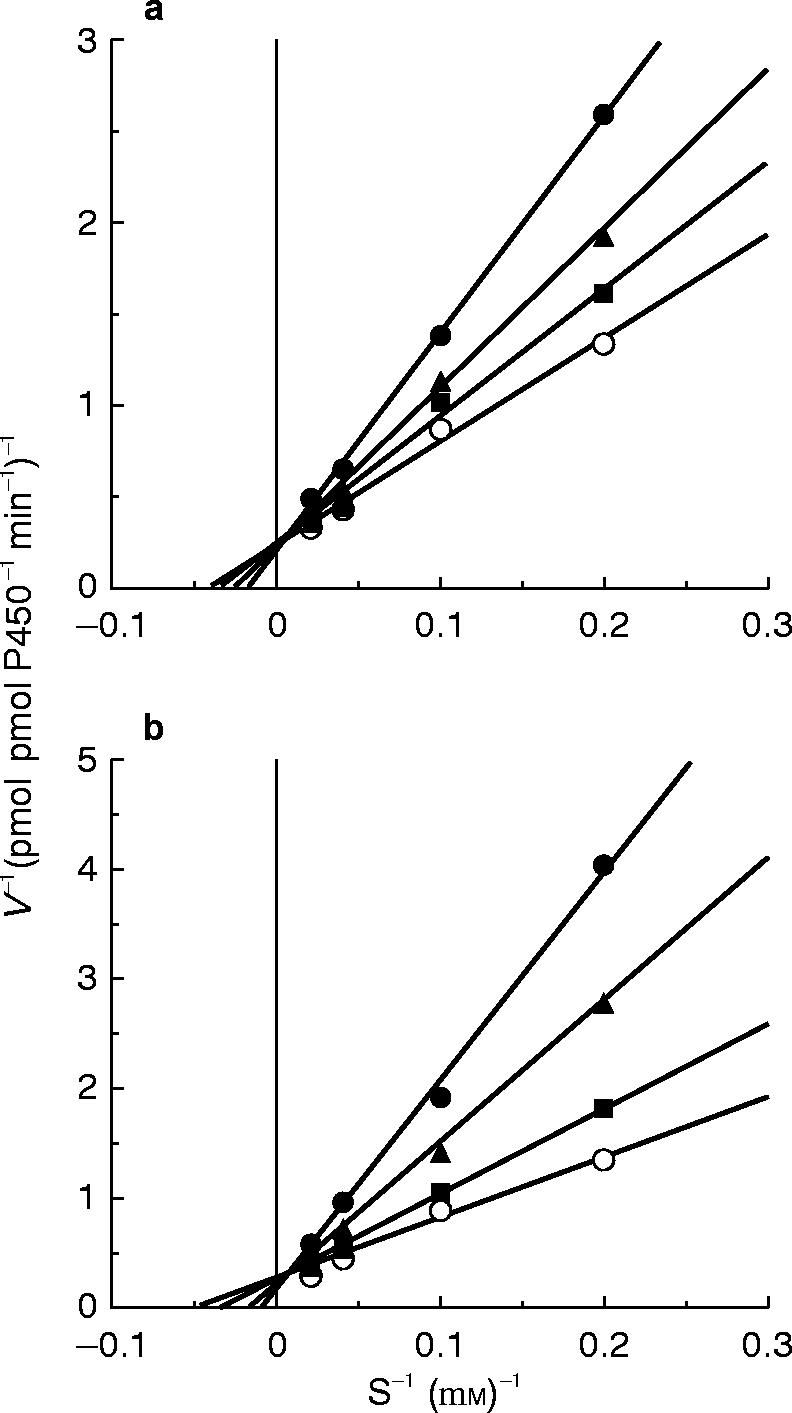
Lineweaver-Burk plots for the inhibition of phenacetin O-deethylation activities by mexiletine (○ 0 μm, ▪ 10 μm, ▴ 25 μm, • 50 μm) (a) and propafenone (○ 0 μm, ▪ 10 μm, ▴ 25 μm, • 50 μm) (b) in microsomes from B-lymphoblastoid cells expressing human CYP1A2. Each data point represents the mean of duplicate determinations.
Discussion
The current in vitro study using human liver microsomes showed that, among eleven antiarrhythmics, mexiletine inhibited phenacetin O-deethylase activity in human liver microsomes with an IC50 value less than 40 μm (Table 1). In addition, we observed that mexiletine selectively abolished the high-affinity component of the phenacetin O-deethylation activity in human liver microsomes (Figure 3b), which is known to be predominantly catalysed by CYP1A2 [6, 8]. Moreover, mexiletine inhibited the phenacetin O-deethylation activity in microsomes of B-lymphoblastoid cells expressing CYP1A2. Taken together, these results strongly suggested that mexiletine is a relatively potent inhibitor of CYP1A2 among the antiarrhythmic drugs studied. Therefore, our in vitro approach using human liver microsomes and recombinant CYP1A2 confirmed that the pharmacokinetic interaction between mexiletine and theophylline, which had been widely documented [19–24], appears to be by inhibition of CYP1A2. Thus, it seems likely that mexiletine has the potential to interact with drugs other than theophylline metabolized by CYP1A2.
We also observed that propafenone selectively abolished the high-affinity component of phenacetin O-deethylation activity in human liver microsomes (Figure 3c) and inhibited phenacetin O-deethylation by recombinant CYP1A2, suggesting that propafenone is also an inhibitor of CYP1A2. A drug-drug interaction between propafenone and theophylline has been reported in only two case reports [36, 37], but little information is available concerning this interaction mechanism. However, our finding suggests that the increase in theophylline concentrations observed after the administration of propafenone [35, 36] may be attributable to the inhibition of CYP1A2 by propafenone.
The inhibition pattern of mexiletine and propafenone for phenacetin O-deethylation in recombinant CYP1A2 was competitive (Figure 4). The results suggest that these drugs bind to the substrate binding site of CYP1A2. This is coincident with the reported finding that the N-demethylation of propafenone was partially catalysed by CYP1A2 [38]. However, the previous in vivo human panel [27, 28] and in vitro human microsomal studies [30, 31] have indicated that the metabolism of mexiletine is mainly mediated by CYP2D6. Nonetheless, the formation rate of hydroxy-methylmexiletine has been reported to be greater in smokers than in nonsmokers, although there was no difference between the two groups in the formation rate of p-hydroxymexiletine [39]. Since cigarette smoking causes the induction of CYP1A2 [8], this CYP isoform may contribute to the formation of the former metabolic process of mexiletine.
The current study showed that the IC50 values of disopyramide, procainamide and pilsicainide were greater than 1 mm (Table 1). Therefore, these drugs may not interfere with the metabolism of theophylline and other drugs metabolized by CYP1A2. In the case of quinidine, flecainide, lignocaine, aprindine and bepridil, their IC50 values were more than 100 μm (Table 1). This is consistent with the observation that flecainide and lignocaine have no effect on the elimination of caffeine, an alternative test probe of CYP1A2 [6], in healthy volunteers [40]. However, in the case of amiodarone, its IC50 value (86 μm) was greater than mexiletine (37 μm) and propafenone (29 μm), despite an observation from a case report [41] that plasma theophylline concentrations in a patient with chronic obstructive pulmonary disease and arrhythmias were doubled after the introduction of amiodarone. Whether the useful class III antiarrhythmic, amiodarone, inhibits the CYP1A2-mediated metabolism of drugs requires further in vivo studies in humans.
Using the kinetic parameters listed in Table 2 for microsomes from three human livers, the percentage of the high-affinity component for phenacetin O-deethylation was expected to be approximately 40%. If both mexiletine and propafenone are selective inhibitors of the high-affinity component, the maximum inhibition by mexiletine or propafenone will be less than 40%. However, these drugs inhibited more than 90% of phenacetin O-deethylation activity in human liver microsomes (Figure 2a). The reason is unknown. Nevertheless, we presume it as a non-specific inhibition of the low-affinity component of phenacetin O-deethylation by mexiletine and propafenone, when the high concentration of the compounds was used. This is because the concentration of mexiletine and propafenone, which showed 90% inhibition of phenacetin O-deethylation, was 1000 μm (Figure 2a). Such a high-concentration of the compounds may cause the non-specific inhibition of the low-affinity enzyme(s). In contrast, the inhibition percentages, when 30 μm mexiletine or propafenone was used, are similar to the maximum inhibition (approximately 40%) estimated from the kinetic data listed in Table 2. These findings suggest that mexiletine and propafenone are relatively specific inhibitors for the high-affinity component of phenacetin O-deethylation when they were used at low concentration (30 μm) as shown in Figure 3.
In the current study using recombinant CYP1A2, both mexiletine and propafenone competitively inhibited the phenacetin O-deethylation with Ki values of 47 and 21 μm, respectively. These Ki values obtained from the recombinant CYP1A2 are higher than those estimated from IC50 values of mexiletine and propafenone in human liver microsomes. This is because, when the Ki values of mexiletine and propafenone in human liver microsomes are calculated for HL-33 with the equation of Ki = IC50Km/(Km+S), they are 6.9 and 5.4 μm, which are one-seventh and one-fourth of the values of the recombinant CYP1A2, respectively. This may suggest that the Ki values of mexiletine and propafenone for phenacetin O-deethylation in human liver microsomes are different from those in the recombinant CYP1A2. There is no data supporting this possibility. However, we observed that the Km values of phenacetin O-deethylation in the recombinant CYP1A2 (12 μm) were 3- to 7-fold higher than those of the high-affinity component in human liver microsomes (Table 2). Although the finding does not necessarily suggest that there is a difference in Ki values of mexiletine and propafenone between the recombinant system and human liver microsomes, the enzymatic character of CYP1A2 may be different between the recombinant system and in human liver microsomes, due to the differences in microsomal lipid and contents of CYP, NADPH-cytochrome P450 reductase and cytochrome b5 [42].
As far as we know, there is no information on free concentrations in liver tissue of antiarrhythmic drugs tested in the present study. We tried to estimate them from the free concentrations of the drugs in plasma, using effective plasma concentration and protein binding of these antiarrhythmic drugs [43]. Except for procainamide, the free concentrations of the drugs in plasma were less than 5 μm, which were lower than the Ki values. However, free concentrations of these drugs in liver tissue may be higher than those in plasma, since they may accumulate in hepatocytes by active transport. Therefore, the lower concentration of free drug in plasma compared with the Ki value does not always rule out the possible occurrence of a drug-drug interaction in vivo.
In conclusion, we have observed that propafenone and mexiletine are relatively potent inhibitors of CYP1A2 among the antiarrhythmic drugs studied. This observation probably explains the mechanism of the in vivo pharmacokinetic interaction between mexiletine and theophylline reported previously [18–23], namely by the inhibition of CYP1A2 catalysing the metabolism of theophylline.
Acknowledgments
This study was supported by a grant-in-aid for Encouragement of Young Scientists from the Ministry of Education (08772172), and grants-in-aid from the Japan Health Science Foundation (1–7-1-C) and Drug Innovation Science Project (1–2-10), Tokyo, Japan.
References
- 1.Nelson DR, Koymans L, Kamataki T, et al. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- 3.Lemoine A, Gautier JC, Azoulay D, et al. Major pathway of imipramine metabolism is catalyzed by cytochrome P-450 1A2 and P-450 3A4 in human liver. Mol Pharmacol. 1993;43:827–832. [PubMed] [Google Scholar]
- 4.Yoshimoto K, Echizen H, Chiba K, Tani M, Ishizaki T. Identification of human CYP isoforms involved in the metabolism of propranolol enantiomers—-N-desisopropylation is mediated mainly by CYP1A2. Br J Clin Pharmacol. 1995;39:421–431. doi: 10.1111/j.1365-2125.1995.tb04472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertilsson L, Carrilo JA, Dahl M-L, et al. Clozapine disposition covaries with CYP1A2 activity determined by a caffeine test. Br J Clin Pharmacol. 1994;38:471–473. doi: 10.1111/j.1365-2125.1994.tb04385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler MA, Iwasaki M, Guengerich FP, Kadlubar FF. Human cytochrome P-450PA (P-450IA2), the phenacetin O-deethylase, is primarily responsible for the hepatic 3-demethylation of caffeine and N-oxidation of carcinogenic arylamines. Biochemistry. 1989;86:7696–7700. doi: 10.1073/pnas.86.20.7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkar MA, Hunt C, Guzelian PS, Karnes T. Characterization of human liver cytochrome P-450 involved in theophylline metabolism. Drug Metab Dispos. 1992;20:31–37. [PubMed] [Google Scholar]
- 8.Sesardic D, Boobis AR, Edwards RJ, Davies DS. A form of cytochrome P450 in man, orthologous to form d in the rat, catalyses the O-deethylation of phenacetin and is inducible by cigarette smoking. Br J Clin Pharmacol. 1988;26:363–372. doi: 10.1111/j.1365-2125.1988.tb03393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walle T, Walle UK, Cowart TD, Conradi EC, Gaffney TE. Selective induction of propranolol metabolism by smoking: additional effects on renal clearance of metabolites. J Pharmacol Exp Ther. 1987;241:928–933. [PubMed] [Google Scholar]
- 10.Haring C, Meise U, Humpel C, et al. Dose-related plasma levels of clozapine: influence of smoking behavior, sex and age. Psychopharmacology. 1989;99:S38–S40. doi: 10.1007/BF00442557. [DOI] [PubMed] [Google Scholar]
- 11.Grygiel JJ, Birkett DJ. Cigarette smoking and theophylline clearance and metabolism. Clin Pharmacol Ther. 1981;30:491–496. doi: 10.1038/clpt.1981.193. [DOI] [PubMed] [Google Scholar]
- 12.Spina E, Campo GM, Avenoso A, et al. Interaction between fluvoxamine and imipramine/desipramine in four patients. Ther Drug Monit. 1992;14:194–196. doi: 10.1097/00007691-199206000-00004. [DOI] [PubMed] [Google Scholar]
- 13.Jerling M, Lindström L, Bondesson U, Bertilsson L. Fluvoxamine inhibition and carbamazepine induction of the metabolism of clozapine: evidence from a therapeutic drug monitoring service. Ther Drug Monit. 1994;16:368–374. doi: 10.1097/00007691-199408000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Jappesen U, Loft S, Poulsen HE, Brøsen K. A fluvoxamine-caffeine interaction study. Pharmacogenetics. 1996;6:213–222. doi: 10.1097/00008571-199606000-00003. [DOI] [PubMed] [Google Scholar]
- 15.Tarrus E, Cami J, Roberts DJ, et al. Accumulation of caffeine in healthy volunteers treated with furafylline. Br J Clin Pharmacol. 1987;23:9–18. doi: 10.1111/j.1365-2125.1987.tb03003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sperber AD. Toxic interaction between fluvoxamine and sustained release theophylline in an 11-year-old boy. Drug Safety. 6:460–462. doi: 10.2165/00002018-199106060-00006. [DOI] [PubMed] [Google Scholar]
- 17.Sesardic D, Boobis AR, Murray BP, et al. Furafylline is a potent and selective inhibitor of cytochrome P450IA2 in man. Br J Clin Pharmacol. 1990;29:651–663. doi: 10.1111/j.1365-2125.1990.tb03686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brøsen K, Skjelbo E, Rasmussen BB, Poulsen HE, Loft S. Fluvoxamine is a potent inhibitor of cytochrome P4501A2. Biochem Pharmacol. 1993;45:1211–1214. doi: 10.1016/0006-2952(93)90272-x. [DOI] [PubMed] [Google Scholar]
- 19.Hurwitz A, Vacek JL, Botteron GW, et al. Mexiletine effects on theophylline disposition. Clin Pharmacol Ther. 1991;50:299–307. doi: 10.1038/clpt.1991.140. [DOI] [PubMed] [Google Scholar]
- 20.Loi C-M, Wei X, Vestal RE. Inhibition of theophylline metabolism by mexiletine in young male and female nonsmokers. Clin Pharmacol Ther. 1991;49:571–580. doi: 10.1038/clpt.1991.67. [DOI] [PubMed] [Google Scholar]
- 21.Stoysich AM, Mohiuddin SM, Destache CJ, Nipper HC, Hilleman DE. Influence of mexiletine on the pharmacokinetics of theophylline in healthy volunteers. J Clin Pharmacol. 1991;31:354–357. doi: 10.1002/j.1552-4604.1991.tb03717.x. [DOI] [PubMed] [Google Scholar]
- 22.Kendall JD, Chrymko MM, Cooper BE. Theophylline-mexiletine interaction: a case report. Pharmacotherapy. 1992;12:416–418. [PubMed] [Google Scholar]
- 23.Stanley R, Comer T, Taylor JL, Saliba D. Mexiletine-theophylline interaction. Am J Med. 1989;86:733–734. doi: 10.1016/0002-9343(89)90463-4. [DOI] [PubMed] [Google Scholar]
- 24.Katz A, Buskila D, Sukenik S. Oral mexiletine-theophylline interaction. Int J Cardiol. 1987;17:227–228. doi: 10.1016/0167-5273(87)90138-0. [DOI] [PubMed] [Google Scholar]
- 25.Huy RH, Jinzhen C, Andreas UF, Ferenc F. Metabolism of theophylline by cDNA-expressed human cytochrome P-450. Br J Clin Pharmacol. 1995;39:321–326. doi: 10.1111/j.1365-2125.1995.tb04455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z-Y, Kaminsky LS. Characterization of human cytochromes P450 involved in theophylline 8-hydroxylation. Biochem Pharmacol. 1995;50:205–211. doi: 10.1016/0006-2952(95)00120-o. [DOI] [PubMed] [Google Scholar]
- 27.Wei X, Loi C-M, Jarvi EJ, Vestal RE. Relative potency of mexiletine, lidocaine, and tocainide as inhibitors of rat liver CYP1A1 activity. Drug Metab Dispos. 1995;23:1335–1338. [PubMed] [Google Scholar]
- 28.Abolfathi Z, Fiset C, Gilbert M, Moerike K, Bélanger PM, Turgeon J. Role of polymorphic debrisoquin 4-hydroxylase activity in the stereoselective disposition of mexiletine in humans. J Pharmacol Exp Ther. 1993;266:1196–1201. [PubMed] [Google Scholar]
- 29.Turgeon J, Fiset C, Giguére R, et al. Influence of debrisoquine phenotype and quinidine on mexiletine disposition in man. J Pharmacol Exp Ther. 1991;259:789–798. [PubMed] [Google Scholar]
- 30.Broly F, Libersa C, Lhermitte M. Mexiletine metabolism in vitro by human liver. Drug Metab Dispos. 1990;18:362–368. [PubMed] [Google Scholar]
- 31.Broly F, Libersa C, Lhermitte M, Dupuis B. Inhibitory studies of mexiletine and dextromethorphan oxidation in human liver microsomes. Biochem Pharmacol. 1990;39:1045–1053. doi: 10.1016/0006-2952(90)90283-q. [DOI] [PubMed] [Google Scholar]
- 32.Chiba K, Manabe K, Kobayashi K, et al. Development and preliminary application of a simple assay of S-mephenytoin 4-hydroxylase in human liver microsomes. Eur J Clin Pharmacol. 1993;44:559–562. doi: 10.1007/BF02440859. [DOI] [PubMed] [Google Scholar]
- 33.Yamaoka K, Tanigawara Y, Nakagawa T, Uno T. A pharmacokinetic analysis program (MULTI) for microcomputer. J Pharmacobio Dyn. 1981;4:879–885. doi: 10.1248/bpb1978.4.879. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi K, Yamamoto T, Chiba K, et al. The effects of selective serotonin reuptake inhibitors and their metabolites on S-mephenytoin 4′-hydroxylase activity in human liver microsomes. Br J Clin Pharmacol. 1995;40:481–485. doi: 10.1111/j.1365-2125.1995.tb05793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevens JC, Wrighton SA. Interaction of the enantiomers of fluoxetine and norfluoxetine with human liver cytochrome P450. J Pharmacol Exp Ther. 1993;266:964–971. [PubMed] [Google Scholar]
- 36.Lee BL, Dohrmann ML. Theophylline toxicity after propafenone treatment: evidence for drug interaction. Clin Pharmacol Ther. 1992;51:353–355. doi: 10.1038/clpt.1992.32. [DOI] [PubMed] [Google Scholar]
- 37.Spinler SA, Gammaitoni A, Charland SL, Hurwitz J. Propafenone-theophylline interaction. Pharmacotherapy. 1993;13:68–71. [PubMed] [Google Scholar]
- 38.Botsch S, Gautier J-C, Beaune P, Eichelbaum M, Kroemer HK. Identification and characterization of the cytochrome P450 enzymes involved in N-dealkylation of propafenone: molecular base for interaction potential and variable disposition of active metabolites. Mol Pharmacol. 1993;43:120–126. [PubMed] [Google Scholar]
- 39.Grech-Bélanger O, Gilbert M, Turgeon J, LeBlanc P-P. Effects of cigarette smoking on mexiletine kinetics. Clin Pharmacol Ther. 1985;37:638–643. doi: 10.1038/clpt.1985.103. [DOI] [PubMed] [Google Scholar]
- 40.Joeres R, Richter E. Mexiletine and caffeine elimination. N Engl J Med. 1987;317:117. doi: 10.1056/NEJM198707093170214. [DOI] [PubMed] [Google Scholar]
- 41.Soto J, Sacristán JA, Arellano F, Hazas J. Possible theophylline-amiodarone interaction. Ann Pharamacother. 1990;24:1115. doi: 10.1177/106002809002401118. [DOI] [PubMed] [Google Scholar]
- 42.Crespi CL. Xenobiotic-metabolizing human cells as tools for pharmacological and toxicological research. In: Testa B, Meyer US, editors. Advances in Drug Research. Vol. 26. London: Academic Press; 1995. pp. 179–235. [Google Scholar]
- 43.Benet LZ, Øie S, Schwartz LB. Design and optimization of dosage regimens; Pharmacokinetic data. In: Harman JG, Limbird LE, Molinoff PB, et al., editors. Goodman & Gilman’s the Pharmacological Basis of Therapeutics. Ninth. New York: McGraw-Hill; pp. 1707–1792. [Google Scholar]