

Abstract
The muscle intracellular (IC) free glucose concentration and the rate of muscle glycogen synthesis were measured by using in vivo 13C and 31P NMR spectroscopy in normal volunteers under hyperinsulinemic (≈300 pM) clamp conditions at the following three plasma glucose levels: euglycemia (≈6 mM), mild (≈10 mM), and high (≈16 mM) hyperglycemia. In keeping with biopsy studies, muscle IC free glucose concentration at euglycemia (−0.03 ± 0.03 mmol/kg of muscle, mean ± SEM, n = 10) was not statistically different from zero. A small but statistically significant amount of IC free glucose was observed during mild and high hyperglycemia: 0.15 ± 0.08 (n = 5) and 0.43 ± 0.20 mmol/kg of muscle (n = 5), respectively. Muscle glycogen synthesis rate, in mmol per kg of muscle per min, was 111 ± 11 at euglycemia (n = 10), 263 ± 29 during mild hyperglycemia (n = 5), and 338 ± 42 during high hyperglycemia (n = 5), these three rates being significantly different from each other. As previous in vitro and in vivo studies, these rates suggest a Km (concentration at which unidirectional glucose transport reaches half-maximal rate) of the muscle glucose transport system in the 15–25 mM range under hyperinsulinemic conditions. The low concentrations of muscle IC free glucose observed under hyperinsulinemic conditions were interpreted, with this estimate and in the framework of metabolic control theory, as glucose transport being the predominant step controlling muscle glucose flux not only at euglycemia but also during hyperglycemia.
Insulin accelerates transmembrane glucose transport in insulin-sensitive tissues, mainly by recruiting GLUT-4 glucose transporters from an intracellular pool (1). Because skeletal muscle has been shown to be the principal site of insulin resistance, transmembrane glucose transport has been proposed as a potential step for causing skeletal muscle insulin resistance (2, 3). It is noteworthy that the mechanism of insulin signaling leading to GLUT-4 recruitment is complex (4), and a defect in any component of this pathway could interfere with glucose transport and insulin sensitivity. The involvement of glucose transport in insulin resistance would be strongly supported if one could determine whether the transport step controls muscle glucose metabolism in all physiological conditions in healthy subjects. In that case, any reduction in the transmembrane glucose transport capacity would be indeed expected to reduce the overall glucose flux. Although transmembrane transport is generally accepted as the dominant step controlling muscle glucose metabolism at euglycemia and physiological insulinemia (5, 6), some step beyond transport might become predominant during hyperglycemic hyperinsulinemia.
Metabolic control theory provides a systematic framework for evaluating quantitatively the control exerted on the flux by an enzymatic step in a pathway (7, 8). In vivo NMR data suggest that the dominant step controlling muscle glucose flux during hyperglycemic hyperinsulinemia in the physiological range is located at the glucose transporter or at the hexokinase step (9). Recent advances in NMR spectroscopy have allowed noninvasive measurements of glucose in human muscle (10, 11). Assessment of intracellular (IC) free glucose would allow discrimination between the roles played by the above-mentioned steps, because the absence of IC free glucose would be a direct evidence of glucose transport being the major flux-controlling step for muscle glucose disposal. We report herein the quantitation of IC free glucose concentration in human muscle by using 13C and 31P NMR spectroscopy. Three different plasma glucose levels were explored during hyperinsulinemic clamps to provide a quantitative estimate of the contribution of glucose transport to the control of muscle glucose flux in normal subjects.
METHODS
Subjects.
Seventeen lean healthy men, aged 19–28 years, with fasting plasma glucose <6 mM and normal glycosylated hemoglobin participated in the protocol. The protocol was approved by the Human Investigation Committee of Kremlin–Bicêtre School of Medicine, and all participants gave written consent after explanation of the purpose, nature, and potential risks of the study. Four experimental groups matched for weight, body mass index, glycosylated hemoglobin, and age were studied. Three subjects participated in two different groups. The characteristics of the groups are summarized in Table 1.
Table 1.
Characteristics of the experimental groups
| Group
|
||||
|---|---|---|---|---|
| I | II | III | IV | |
| n | 5 | 5 | 5 | 5 |
| Age, years | 24 ± 1 | 23 ± 1 | 22 ± 1 | 22 ± 1 |
| Weight, kg | 67 ± 2 | 70 ± 2 | 67 ± 2 | 69 ± 3 |
| BMI, kg/m2 | 22.2 ± 0.6 | 23.3 ± 0.7 | 22.1 ± 0.9 | 21.7 ± 1.3 |
| HbA1c, %* | 4.8 ± 0.2 | 5.2 ± 0.2 | 5.0 ± 0.2 | 5.2 ± 0.2 |
| Fasting glucose, mM | 4.8 ± 0.2 | 5.8 ± 0.3 | 5.4 ± 0.2 | 5.2 ± 0.2 |
| Fasting insulin, pM | 27 ± 4 | 40 ± 4 | 39 ± 3 | 33 ± 3 |
| Hyperglycemic goal, mM | 10 | 10 | 16 | 16 |
| NMR spectroscopy | 13C | 31P | 13C | 31P |
Results are expressed as the mean ± SEM. BMI, body mass index; HbA1c, glycosylated hemoglobin.
Normal range in our laboratory is 4.8–6.4%.
Experimental Protocol.
The experimental protocol consisted of two glucose clamps; the first at a glucose level of ≈10 mM (groups I and II) or ≈16 mM (groups III and IV) lasted 2 h and was followed by a euglycemic clamp lasting 1 h, performed in all groups. Because acquiring 31P and 13C NMR data simultaneously was not technically feasible on our spectrometer, we performed these measurements separately in different groups. Thus groups I and III were examined by 13C NMR spectroscopy and groups II and IV were examined by 31P NMR spectroscopy (see Table 1). Studies were started at the hour of 0730 after 11–13 h of fasting. A Teflon catheter was inserted into a vein of each forearm for blood drawing and glucose/hormone infusion. After a 15-min basal sampling period, an infusion of somatostatin (Modustatine, Sanofi, Gentilly; 0.1 μg per min per kg of body weight) was initiated at time −10 min and continued throughout the study to inhibit endogenous insulin secretion. At time 0 min, a primed continuous infusion of insulin (Actrapid, Novo-Nordisk, Copenhagen) was started (6 pmol per min per kg of body weight) and plasma glucose was raised to the hyperglycemic goal (either ≈10 or ≈16 mM) by a rapid infusion of 1.11 M glucose. For groups I and III only, 4 g of [1-13C]glucose (Isotec) was also infused during the initial bolus. The hyperglycemic plateau was maintained by means of a variable rate intravenous infusion of 1.11 M glucose (unlabeled for groups II and IV, 15% and 10% [1-13C]glucose for groups I and III, respectively). Plasma glucose concentration was determined at 5-min intervals from arterialized blood and the glucose infusion rate was automatically adjusted by a microcomputer by using algorithms as described (12). At 120 min, the glucose infusion was interrupted, and plasma glucose concentration was allowed to decrease to ≈6 mM. The glucose clamp was then resumed for 1 h at euglycemia (unlabeled glucose for groups II and IV, 34% [1-13C]glucose for groups I and III). The change in the 13C enrichment of the glucose infusate in groups I and III was designed to limit the variation in signal-to-noise ratio of the glucose 13C NMR C1 resonances throughout the study. Arterialized blood samples were taken every 15 min for measurement of plasma insulin and glucose 13C enrichment (groups I and III only).
NMR Spectroscopy.
During the measurements, the subjects remained supine within an NMR spectrometer (Bruker; 72-cm free bore, 3 T). For subjects in groups I and III, the right leg was placed inside a 14-cm diameter linear bird-cage tuned to the 13C frequency and centered on the largest calf diameter. The length of the 13C volume coil was 14 cm; it was surrounded by a slightly larger linear birdcage tuned to the 1H frequency. All 13C NMR spectra were recorded with full nuclear Overhauser enhancement but without 1H decoupling. The pulse sequences and the absolute quantitation method used for muscle glucose concentration measurements have been described in detail (11). Shortly, the [1-13C]glucose signal was collected by using a spin-echo sequence with TE = 15 ms to reduce the large glycogen signal, overlapping the glucose signal at the end of the hyperglycemic period (see Fig. 2). This signal was quantitated by comparison to a calibration glucose phantom with the appropriate corrections for partial saturation, T2 attenuation, probe loading, and sensitive volume geometry. For glycogen synthesis rate measurements, additional pulse-acquire spectra with TR = 400 ms were acquired. The increment in [1-13C]glycogen concentration was determined from the signal obtained by difference of two successive spectra. This signal was quantitated by comparison to a calibration glycogen phantom by using the same corrections that were used for probe loading and sensitive volume geometry.
Figure 2.

13C NMR spectra (110- to 85-ppm region) of the human muscle obtained during the second hour of a hyperglycemic hyperinsulinemic clamp for a subject in group III. Upper curve, pulse-acquire spectrum with TR = 400 ms (3,000 scans); lower curve, spin-echo spectrum with TE = 15 ms and TR = 1.2 s (1,000 scans). The dramatic reduction of the glycogen signal facilitates glucose signal quantitation.
For subjects in groups II and IV, 31P NMR spectra were obtained with a 5-cm diameter surface coil placed under the calf, by using a pulse-acquire sequence with TR = 2.4 s. Full nuclear Overhauser enhancement and decoupling were obtained using a larger 1H coil. The flip angle was adjusted to maximize the phosphocreatine signal. Concentrations of glucose 6-phosphate (G6P) were calculated from the data as described (2, 13) by using the β-ATP resonance as an internal concentration standard assumed to represent a concentration of 5.5 mmol/kg of muscle (14).
Analytical Procedures.
Plasma glucose concentration was measured by the glucose oxidase method (Beckman) and plasma insulin by radioimmunoassay (Pharmacia). Hematocrit was determined by conductivity (Nova). Plasma glucose 13C enrichment was determined by high-resolution 1H NMR after deproteinization, lyophilization, and redissolution in 2H2O.
Calculations.
13C NMR data were derived from groups I and III, and 31P NMR data were derived from groups II and IV. To calculate the IC free glucose concentration in the three glycemic conditions, group I was assumed to behave identically to group II and group III was assumed to behave identically to group IV. The NMR-determined [1-13C]glucose concentration was converted to the total concentration of IC and extracellular (EC) glucose plus G6P by using the plasma glucose 13C enrichment measured simultaneously. The total free glucose concentration (EC + IC) was thus derived by subtracting the G6P concentration, measured independently by 31P NMR, from the 13C NMR result. EC glucose muscle content was calculated from the plasma glucose concentration by assuming that resting muscle contains 70 ml of EC water per kg of wet tissue (15). Glycogen synthesis rates were calculated from the increment in [1-13C]glycogen concentration and the plasma glucose 13C enrichment determined over the same time period (16).
Data Analysis.
Data are expressed as the mean ± SEM. Blood variables, muscle G6P and glucose concentrations, and muscle glycogen synthesis rate are given as the average of the last 45 min of the hyperglycemic or euglycemic period. Results were analyzed by one-way or two-way ANOVA, with or without repeated measurements, where appropriate. Significance was set at P < 0.05. When needed, honestly significant difference Tukey multiple comparison of means was performed.
RESULTS
Table 2 shows plasma glucose and insulin values and the hematocrits during the clamps. Stable and comparable plasma insulin levels (≈300 pM) were obtained in all four groups during the studies (difference between groups, F = 1.92 and P = 0.166; difference between plasma glucose levels, F = 0.00, P = 0.993). Plasma glucose levels were not significantly different in the 13C and 31P groups that were studied at a mild hyperglycemic plateau (groups I and II, P = 0.552 at euglycemia and P = 1.000 during hyperglycemia) nor in the 13C and 31P groups that were studied at the high hyperglycemic plateau (groups III and IV, P = 0.349 at euglycemia and P = 0.251 during hyperglycemia). The hematocrits were stable throughout the experiments (difference between groups, F = 0.49, P = 0.504; difference between plasma glucose levels, F = 2.78, P = 0.134).
Table 2.
Average blood variables during the hyperglycemic and euglycemic clamps for groups I–V
| Clamp | Group
|
||||
|---|---|---|---|---|---|
| I | II | III | IV | ||
| Plasma glucose, mM | Hyperglycemia | 9.8 ± 0.1 | 9.8 ± 0.1 | 16.4 ± 0.2 | 17.8 ± 1.0 |
| Euglycemia | 5.5 ± 0.1 | 5.7 ± 0.1 | 5.8 ± 0.1 | 6.1 ± 0.2 | |
| Plasma insulin, pM | Hyperglycemia | 318 ± 15 | 274 ± 6 | 301 ± 15 | 292 ± 24 |
| Euglycemia | 308 ± 15 | 265 ± 17 | 323 ± 14 | 289 ± 25 | |
| Hematocrit, % | Hyperglycemia | 41.5 ± 0.2 | 41.5 ± 0.2 | 41.4 ± 1.3 | 40.9 ± 1.4 |
| Euglycemia | 41.5 ± 0.6 | 41.2 ± 0.6 | 42.2 ± 1.3 | 42.3 ± 1.5 | |
Fig. 1 shows 31P NMR spectra obtained from a subject during the hyperglycemic and euglycemic periods. In the difference spectrum, the increase in G6P and inorganic phosphate and the decrease in phosphocreatine could be clearly seen. On average, the G6P concentration assessed by 31P NMR were significantly higher during the hyperglycemic clamp, as compared with the euglycemic period (F = 90.66, P < 0.001): 200 ± 15 vs. 149 ± 11 μmol/kg of muscle in group II and 233 ± 16 vs. 157 ± 19 μmol/kg of muscle in group IV. Fig. 2 shows 13C NMR spectra obtained from a subject at the end of the hyperglycemic period, using the pulse-acquire sequence and the short spin-echo sequence. Due to the significant attenuation of the glycogen signal at TE = 15 ms, the glucose signal could be clearly observed in spite of the large glycogen accumulation.
Figure 1.

31P NMR spectra of the human muscle obtained during the euglycemic (top trace) and hyperglycemic (middle trace) clamps for a subject in group IV. In the difference spectrum (bottom trace, hyperglycemia minus euglycemia), an increase in G6P and inorganic phosphate (Pi) and a decrease in phosphocreatine (PCr) are observed. In the spectrum obtained at euglycemia, the G6P concentration was determined by integrating from 7.43 to 7.13 ppm and multiplying the area by 2 (2, 13). The G6P resonance intensity in the difference spectrum corresponds to a concentration difference of 88 μmol/kg of muscle.
The muscle content in G6P, EC glucose, and IC free glucose is depicted in Fig. 3 for the three glycemic conditions studied: euglycemia and mild and high hyperglycemia. For each glycemia, total muscle glucose (IC + EC) plus G6P concentration was obtained through the 13C NMR measurements (groups I and III), muscle G6P concentration was determined by 31P NMR (groups II and IV), and muscle EC glucose concentration was estimated from plasma glucose levels. Muscle IC free glucose concentration was computed by subtracting the last two components (G6P and EC glucose) from the first one (IC + EC glucose plus G6P). Because the experimental errors are largely dominated by the 13C NMR measurements, the variability in muscle IC free glucose is numerically very close to that in total muscle glucose plus G6P. For the euglycemic condition, data from groups I and III, on one hand, and groups II and IV, on the other hand, were pooled. No IC free glucose was detected at euglycemia. The negative value plotted in Fig. 3 simply reflects that the total concentration in muscle glucose plus G6P, as detected by 13C NMR, was on average slightly lower than the concentration anticipated from an EC water fraction of 7%. The concentration in muscle glucose plus G6P increased significantly from euglycemia to hyperglycemia (F = 127.07, P < 0.001) and was higher during high hyperglycemia as compared with mild hyperglycemia (interaction between group and glycemic level, F = 14.42 and P = 0.005). This variation was partly due to the contribution from EC glucose. More interestingly, a statistically significant accumulation of IC free glucose was detected in both hyperglycemic conditions as compared with euglycemia (F = 11.50, P = 0.009). When a linear regression was used on individual data, muscle IC free glucose concentration appeared positively correlated to the glycemia (r = 0.66 and P = 0.001).
Figure 3.

Total muscle glucose (IC + EC) plus G6P concentration during hyperinsulinemia at three glycemic levels. G6P, EC glucose, and IC glucose contributions are represented by stippled, hatched, and open bars, respectively. The error bars are given for IC glucose and are numerically very close to the error bars for total glucose plus G6P.
As shown in Fig. 4, muscle glycogen synthesis rate was highly correlated to the glucose infusion rate, indicating that the variability in the glycogen synthesis rates was mainly related to the variability among subjects rather than to experimental errors. Glycogen synthesis rate, in μmol per kg of muscle per min, was 111 ± 11 at euglycemia, 263 ± 29 during mild hyperglycemia, and 338 ± 42 during high hyperglycemia. Individual data are presented in Fig. 5. As compared with euglycemia, glycogen synthesis rate was significantly increased in both hyperglycemic conditions (F = 83.54 and P < 0.001). Glycogen synthesis rate at the high hyperglycemic plateau was also significantly higher than at the mild hyperglycemic plateau (interaction between group and glycemic level, F = 5.96 and P = 0.040). When a linear regression was used on individual data, muscle glycogen synthesis rate appeared positively correlated to glycemia (r = 0.84 and P < 0.001).
Figure 4.

Muscle glycogen synthesis rate as a function of glucose infusion rate for the experiments in groups I and III. The dashed line is the linear best fit of the data (r = 0.92 and P < 0.001).
Figure 5.

Muscle glycogen synthesis rate as a function of plasma glucose level for the experiments in groups I and III.
DISCUSSION
Previous human studies have shown that muscle IC glucose content at euglycemia remains beneath the sensitivity limit of the muscle biopsy technique (6, 17–19), even in the presence of high physiological plasma insulin. Our results obtained by in vivo NMR spectroscopy are consistent with this well-established feature, interpreted as transmembrane glucose transport being the dominant step controlling muscle glucose metabolism at euglycemia. Biopsy as well as in vivo NMR studies (10, 20) performed in hyperglycemic conditions with basal insulin have demonstrated a moderate accumulation of IC free glucose in human muscle, suggesting that some step beyond transport might participate to the control of glucose flux in this nonphysiological situation. The present study examined two hyperinsulinemic (≈300 pM) hyperglycemic conditions: a mild hyperglycemic plateau (glycemia ≈10 mM) and a high hyperglycemic plateau (glycemia ≈16 mM). In both conditions, a low but statistically significant amount of IC free glucose could be detected. Because of the low content in IC free glucose, the correction of the 13C NMR muscle glucose signal for the G6P contribution appeared to be critical. However, in quantitative agreement with previous in vivo NMR studies (21), our 31P NMR data demonstrate a moderate increase in G6P between euglycemia and hyperglycemia under hyperinsulinemic conditions. By using the formalism of metabolic control analysis (7, 9), the limited rise in muscle G6P concentration with glycemia can be interpreted as reflecting a high elasticity coefficient of glycogen synthase with respect to G6P, because the proportionality coefficient between glycogen synthesis rate and plasma glucose level is equal or higher than unity. This feature supports the recently addressed concept of maintenance of G6P homeostasis (22). Another possible error inducing an artifactual appearance of muscle IC free glucose could be the increase of the EC volume, induced by the hyperglycemia. The stability of the hematocrit during the experiments indicates that no detectable water shift from the IC to the EC space occurred.
Under the assumption that resting muscle contains 700 ml of IC water per kg of wet tissue (15), the IC free glucose concentrations determined during ≈10 mM and ≈16 mM hyperglycemia can be converted into EC-to-IC glucose gradients of 46:1 and 26:1, respectively. These large gradients suggest that transmembrane transport remains the dominant flux-controlling step in both hyperglycemic hyperinsulinemic conditions. However, a quantitative estimate of the contribution of glucose transport to the control of muscle glucose flux can be obtained using a symmetric Michaelis–Menten kinetic model in the framework of metabolic control theory (see Appendix). This simple kinetic model was chosen because glucose carrier molecular mechanism is not sufficiently characterized to definitely support a more sophisticated description. However, using a kinetic model accounting for product inhibition (7) would provide quantitative estimates very similar to those presented below. As shown in Appendix, the control coefficient of the transport step (CT) can be expressed as the ratio of the variation of the net glucose flux between two different glycemias to the variation of the inward glucose flux. Because EC and IC muscle glucose concentrations are available at three glycemias, CT can be calculated for the mild hyperglycemic range (6–10 mM) and the high hyperglycemic range (10–16 mM) by replacing the differential variations of EC and IC glucose concentrations, in Eq. 8 of Appendix, by the finite variations observed experimentally. This calculation also requires an estimate of the Km (concentration at which unidirectional glucose transport reaches half-maximal rate) for the muscle glucose transport system under hyperinsulinemic conditions. Using a single value of Km for the whole glycemic range appears valid because insulinemia was comparable in the different glycemic conditions studied and because short-term hyperglycemia is not expected to modulate glucose transport activity. Recent in vitro studies on the GLUT-4 transporter (23–25), as well as whole body and forearm glucose uptakes measured in hyperinsulinemic conditions (26), have provided consistent Km values in the 20 mM range. A Km of 17 mM can be estimated from the present IC and EC glucose data with Eq. 5 of Appendix to describe glucose transport kinetics and taking the observed glycogen synthesis rates as an approximation for muscle glucose uptake during hyperglycemic hyperinsulinemia (16). This Km value is also consistent with the glycogen synthesis rate observed at euglycemia, if in this hyperinsulinemic condition, glycogen synthesis represents 50–60% of muscle glucose uptake (21, 27, 28). Thus, for the mild hyperglycemic range, the calculated CT value falls between 0.89 ± 0.08 and 0.91 ± 0.06 for estimates of Km ranging from 15 to 25 mM. In other words, whatever the value of Km, the present data demonstrate that about 90% of the glucose flux control is exerted by the transmembrane transport in mild hyperinsulinemic hyperglycemic conditions. Similarly, in the high hyperglycemic range, the calculated CT falls between 0.80 ± 0.16 and 0.86 ± 0.11 for estimates of Km ranging from 15 to 25 mM. In this situation, where the glycemia approaches Km, a less accurate estimate of CT is obtained, but transmembrane glucose transport still appears as the dominant flux-controlling step.
Our finding that transmembrane glucose transport almost entirely exerts the control of glucose flux in human skeletal muscle, not only at euglycemia but also in hyperglycemic hyperinsulinemic conditions, is in keeping with several studies on laboratory animals (5, 29). By using glycogen synthesis rates and G6P concentrations measured in humans by in vivo NMR, Shulman et al. (9) have reached conclusions consistent with our conclusions, but the distinction between the glucose transporter and the hexokinase step was not possible in the absence of muscle IC free glucose data. Thus, the in vivo NMR measurement of IC free glucose appears as a promising alternate to the biopsy technique for specifically studying glucose transport in healthy subjects and in insulin-resistant patients. However, although in vivo NMR spectroscopy and needle biopsy provide consistent data, as shown at euglycemia in this study, a common critique to both techniques is that the IC volume of distribution for glucose could be smaller than the IC water space. As a consequence, the IC glucose concentrations estimated by these methods would be underestimated. In a recent study based on a new three-tracer technique, Saccomani et al. (30) used a sophisticated multicompartment kinetic model to estimate muscle glucose transmembrane transport and phosphorylation, as well as EC and IC glucose distribution volumes and concentrations, in the forearm of healthy subjects. During physiological hyperinsulinemia, the estimated IC/EC ratio of glucose distribution volumes was only 6.2, as compared with 10 for water (15). Hence, the IC glucose concentration evaluated at euglycemia was 0.69 ± 0.15 mmol/liter of glucose distribution volume. This discrepancy illustrates that the estimation of the true distribution volumes of glucose is critical for the calculation of local glucose concentrations, whatever the method used. Although more experimental evidence about muscle IC compartmentation will be necessary to decide whether the assumptions generally accepted in biopsy studies need to be revised, it was interesting to reexamine the present data by using the IC glucose distribution volume estimated under hyperinsulinemic conditions in (30). By doing so, we found that the IC glucose concentrations increased by a factor of 1.6 so that the EC-to-IC gradients decreased to 29:1 and 16:1 during mild and high hyperglycemia, respectively. As a consequence, the calculated values of CT were slightly decreased (0.82 ± 0.07 to 0.86 ± 0.06 during mild hyperglycemia, and 0.68 ± 0.16 to 0.78 ± 0.11 during high hyperglycemia, for estimates of Km ranging from 15 to 25 mM, respectively), but glucose transport still appears as the predominant flux-controlling step for muscle glucose metabolism, especially in the mild hyperglycemic range. The fact that CT remains close to 1 for a large range of assumed values of the IC glucose distribution volume, as well as of Km, reflects the very small increase in IC free glucose as compared with the increment in EC glucose concentration. Indeed, as evidenced in Eq. 8 of Appendix, the difference of CT from unity is directly proportional to the ratio of the increase in IC free glucose to the increase in EC glucose.
The ratio of the glucose phosphorylation flux to the inward transmembrane glucose flux (Fmet/Fin), measured by a tracer technique, has been proposed by different groups (29, 30) to evaluate the role of glucose transport as a control step of muscle glucose metabolism. Although this ratio does not rigorously correspond to the control coefficient of glucose transport, a value close to unity is indicative of glucose transport being the dominant flux-controlling step. From the results in ref. 30, based on the washout curves of tracers in venous blood, Fmet/Fin can be estimated to 0.36 ± 0.19 in the basal state and to 0.67 ± 0.17 during euglycemic hyperinsulinemia. These results are in contradiction to the widely held opinion that glucose transport is the flux-controlling step of muscle glucose metabolism at euglycemia. However, this discrepancy is not a general feature of tracer approaches. Another tracer study in the euglycemic anesthetized rat (29) has reported values of Fmet/Fin much closer to unity: 0.96 ± 0.05 and 0.78 ± 0.15 in the basal state, 0.90 ± 0.02 and 0.94 ± 0.01 during euglycemic hyperinsulinemia, for red and white muscle, respectively. It is noteworthy that these results were obtained by in situ freeze-clamping of the muscle and that, in spite of anesthesia, measured muscle glucose uptakes were higher than those observed in human muscle in ref. 30. It seems beyond the scope of this paper to compare the validity of the tracer models proposed by Saccomani et al. (30) and by Furler et al. (29). The difficulty to quantitate metabolic fluxes through indirect measurements, such as those described in ref. 30, is illustrated by the large discrepancy between the values of Fmet and of the forearm glucose uptake (obtained by arterio-venous difference) in the basal state: 8.4 ± 2.2 versus 3.3 ± 1.2 μmol per kg of muscle per min. However, this limitation could be circumvented by measuring fractional changes in Fin and Fmet in response to a small change in glycemia. This approach would reduce the bias due to possible systematic errors in tracer modeling and would provide results directly interpretable in the rigorous framework of metabolic control theory.
CONCLUSION
We have demonstrated that in vivo NMR permits direct quantitation of muscle glucose in humans. Under the same assumptions used in biopsy studies, IC free glucose concentration can be calculated from NMR-determined muscle glucose content. The very low concentrations of IC free glucose observed during hyperinsulinemia at different glycemic levels can be interpreted as glucose transport being the predominant step controlling muscle glucose flux for a wide range of physiological conditions in healthy subjects. Similar investigations in subjects with variable degrees of insulin sensitivity are needed to assess the implication of glucose transport in the pathophysiology of muscle insulin resistance.
ABBREVIATIONS
- G6P
glucose 6-phosphate
- IC
intracellular
- EC
extracellular
Appendix
In the framework of metabolic control analysis (7, 8), the control coefficient of glucose transport is defined as the fractional change in the pathway flux F for a fractional change in the concentration of the transporter T
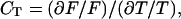 |
1 |
where the partial derivatives indicate that all other enzyme activities are held constant. A flux control coefficient of unity would indicate that glucose uptake is proportional to the activity of the transporter. Because this activity cannot be modulated independently in vivo, another experimental evaluation of CT must be used. Kacser and Burns (7) have demonstrated that CT can be calculated as
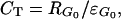 |
2 |
where RG0 is defined as the response coefficient of the flux to EC glucose (concentration G0)
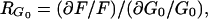 |
3 |
and ɛG0 is defined as the elasticity coefficient of the glucose transporter to EC glucose.
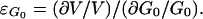 |
4 |
In Eq. 4, ∂V/V is the fractional change in the transporter velocity under in vivo conditions for an isolated variation of G0. On the other hand, in Eq. 3, ∂F/F is the fractional change in the in vivo glucose flux, taking into account the possible variation of IC free glucose concentration (Gi). When a symmetric Michaelis–Menten kinetics are assumed, F (or V) can be written as
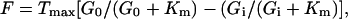 |
5 |
where Tmax is the maximum unidirectional glucose transport rate. Substituting Eq. 5 into Eqs. 3 and 4 gives
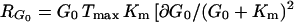 |
6 |
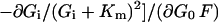 |
and
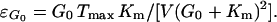 |
7 |
If F = V, Eq. 2 can be reexpressed as
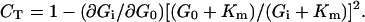 |
8 |
Thus, CT can be experimentally evaluated from Gi measurements at two different values of G0, provided an estimate of Km is known. An expression identical to Eq. 8 is obtained more intuitively by defining CT(G0) as the ratio of the change in the pathway flux F to the change of the inward flux Fin in response to a change in G0
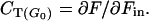 |
9 |
References
- 1.Barnard R J, Youngren J F. FASEB J. 1992;6:3238–3244. doi: 10.1096/fasebj.6.14.1426762. [DOI] [PubMed] [Google Scholar]
- 2.Rothman D L, Shulman R G, Shulman G I. J Clin Invest. 1992;89:1069–1075. doi: 10.1172/JCI115686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonadonna R C, Del Prato S, Saccomani M P, Bonora E, Gulli G, Ferrannini E, Bier D, Cobelli C, DeFronzo R A. J Clin Invest. 1993;92:486–494. doi: 10.1172/JCI116592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White M F, Kahn C R. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- 5.Ziel F H, Venkatesan N, Davidson M B. Diabetes. 1988;37:885–890. doi: 10.2337/diab.37.7.885. [DOI] [PubMed] [Google Scholar]
- 6.Katz A, Nyomba B L, Bogardus C. Am J Physiol. 1988;255:E942–E945. doi: 10.1152/ajpendo.1988.255.6.E942. [DOI] [PubMed] [Google Scholar]
- 7.Kacser H, Burns J A. Symp Soc Exp Biol. 1973;32:65–104. [PubMed] [Google Scholar]
- 8.Fell D A. Biochem J. 1992;286:313–330. doi: 10.1042/bj2860313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shulman R G, Bloch G, Rothman D L. Proc Natl Acad Sci USA. 1995;92:8535–8542. doi: 10.1073/pnas.92.19.8535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roussel R, Velho G, Carlier P G, Jouvensal L, Bloch G. Am J Physiol. 1996;271:E434–E438. doi: 10.1152/ajpendo.1996.271.3.E434. [DOI] [PubMed] [Google Scholar]
- 11.Roussel R, Carlier P G, Wary C, Velho G, Bloch G. Magn Reson Med. 1997;37:821–824. doi: 10.1002/mrm.1910370604. [DOI] [PubMed] [Google Scholar]
- 12.DeFronzo R A, Tobin J E, Andres R. Am J Physiol. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 13.Bloch G, Chase J R, Avison M J, Shulman R G. Magn Reson Med. 1993;30:347–350. doi: 10.1002/mrm.1910300311. [DOI] [PubMed] [Google Scholar]
- 14.Harris R C, Hultman E, Nordesjo L O. Scand J Clin Lab Invest. 1974;33:109–120. [PubMed] [Google Scholar]
- 15.Sjøgaard G, Saltin B. Am J Physiol. 1982;243:R271–R280. doi: 10.1152/ajpregu.1982.243.3.R271. [DOI] [PubMed] [Google Scholar]
- 16.Shulman G I, Rothman D L, Jue T, Stein P, DeFronzo R A, Shulman R G. N Engl J Med. 1990;322:223–228. doi: 10.1056/NEJM199001253220403. [DOI] [PubMed] [Google Scholar]
- 17.Yki-Järvinen H, Sahlin K, Ren J M, Koivisto V A. Diabetes. 1990;39:157–167. doi: 10.2337/diab.39.2.157. [DOI] [PubMed] [Google Scholar]
- 18.Vaag A, Henriksen J E, Beck-Nielsen H. J Clin Invest. 1992;89:782–788. doi: 10.1172/JCI115656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schalin J C, Harkonen M, Groop L C. Diabetes. 1992;41:598–604. doi: 10.2337/diab.41.5.598. [DOI] [PubMed] [Google Scholar]
- 20.Katz A, Raz I, Spencer M K, Rising R, Mott D M. Am J Physiol. 1991;260:R698–R703. doi: 10.1152/ajpregu.1991.260.4.R698. [DOI] [PubMed] [Google Scholar]
- 21.Rothman D L, Magnusson I, Cline G, Gerard D, Kahn C R, Shulman R G, Shulman G I. Proc Natl Acad Sci USA. 1995;92:983–987. doi: 10.1073/pnas.92.4.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shulman R G, Rothman D L. Proc Natl Acad Sci USA. 1996;93:7491–7495. doi: 10.1073/pnas.93.15.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodyear L J, Hirshman M F, Smith R J, Horton E S. Am J Physiol. 1991;261:E556–E561. doi: 10.1152/ajpendo.1991.261.5.E556. [DOI] [PubMed] [Google Scholar]
- 24.Ploug T, Galbo H, Ohkuwa T, Tranum-Jensen J, Vinten J. Am J Physiol. 1992;262:E700–E711. doi: 10.1152/ajpendo.1992.262.5.E700. [DOI] [PubMed] [Google Scholar]
- 25.Ploug T, Wojtaszewski J, Kristiansen S, Hespel P, Galbo H, Richter E A. Am J Physiol. 1993;264:E270–E278. doi: 10.1152/ajpendo.1993.264.2.E270. [DOI] [PubMed] [Google Scholar]
- 26.Yki-Järvinen H, Young A A, Lamkin C, Foley J E. J Clin Invest. 1987;79:1713–1719. doi: 10.1172/JCI113011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelley D E, Mandarino L J. J Clin Invest. 1990;86:1999–2007. doi: 10.1172/JCI114935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rossetti L, Lee Y T, Ruiz J, Aldridge S C, Shamoon H, Boden G. Am J Physiol. 1993;265:E761–E769. doi: 10.1152/ajpendo.1993.265.5.E761. [DOI] [PubMed] [Google Scholar]
- 29.Furler S M, Jenkins A B, Storlien L H, Kraegen E W. Am J Physiol. 1991;261:E337–E347. doi: 10.1152/ajpendo.1991.261.3.E337. [DOI] [PubMed] [Google Scholar]
- 30.Saccomani M P, Bonadonna R C, Bier D M, DeFronzo R A, Cobelli C. Am J Physiol. 1996;270:E170–E185. doi: 10.1152/ajpendo.1996.270.1.E170. [DOI] [PubMed] [Google Scholar]