

Abstract
The Ewing’s sarcoma (ES) family of tumors, including peripheral neuroectodermal tumor (PNET), is defined genetically by specific chromosomal translocations resulting in fusion of the EWS gene with a member of the ETS family of transcription factors, either FLI1 (90–95%) or ERG (5–10%). A second level of molecular genetic heterogeneity stems from the variation in the location of the translocation breakpoints, resulting in the inclusion of different combinations of exons from EWS and FLI1 (or ERG) in the fusion products. The most common type of EWS-FLI1 fusion transcript, type 1, is associated with a favorable prognosis and appears to encode a functionally weaker transactivator, compared to other fusion types. We sought to determine whether the observed covariation of structure, function, and clinical course correlates with tumor cell kinetic parameters such as proliferative rate and apoptosis, and with expression of the receptor for insulin-like growth factor I (IGF-1R). In a group of 86 ES/PNET with defined EWS-ETS fusions (45 EWS-FLI1 type 1, 27 EWS-FLI1 non-type 1, 14 EWS-ERG), we assessed proliferation rate by immunostaining for Ki-67 using MIB1 antibody (n = 85), apoptosis by TUNEL assay (n = 66), and IGF-1R expression by immunostaining with antibody 1H7 (n = 78). Ki-67 proliferative index was lower in tumors with EWS-FLI1 type 1 than those with non-type 1 EWS-FLI1, whether analyzed as a continuous (P = 0.049) or categorical (P = 0.047) variable. Logistic regression analysis suggests that this association was secondary to the association of type 1 EWS-FLI1 and lower IGF-1R expression (P = 0.04). Comparing EWS-FLI1 to EWS-ERG cases, Ki-67 proliferative index was higher in the latter (P = 0.01, Mann-Whitney test; P = 0.02, Fisher’s exact test), but there was no significant difference in IGF-1R. TUNEL results showed no significant differences between groups. Our results suggest that clinical and functional differences between alternative forms of EWS-FLI1 are paralleled by differences in proliferative rate, possibly mediated by differential regulation of the IGF-1R pathway.
The Ewing’s sarcoma (ES) family of tumors, including peripheral neuroectodermal tumor (PNET), represents a clinicopathological entity with a variable neural differentiation, usually appearing as a bone or soft tissue lesion in a child or young adult. The primary genetic event is chromosomal translocation resulting in fusion of the EWS gene with a member of the ETS family of transcription factors. 1 The most frequent translocation partner of EWS is FLI1 (90–95%), followed by ERG (5–10%). These fusion products function as oncogenic aberrant transcription factors. 2 Detection of these fusions is considered to be specific for ES/PNET, and has become a valuable tool for the differential diagnosis of primitive small round cell tumors. 3,4
There is a considerable molecular genetic heterogeneity within ES/PNET. As mentioned above, either FLI1 or ERG can rearrange with EWS in these gene fusions. Furthermore, for either gene fusion, additional heterogeneity stems from the location of the genomic breakpoints of the translocation, resulting in different combinations of exons from EWS and FLI1 (or ERG). 5 The most common fusion joins EWS exon 7 in frame with FLI1 exon 6 (type 1 fusion). There are at least 12 other EWS-FLI1 types, and at least 4 types of EWS-ERG fusion types described. We and others 6 have previously reported that among patients with EWS-FLI1 fusions, those having type 1 transcripts have a better outcome regardless of conventional prognostic factors such as stage, age, or tumor location. 7
The biological mechanisms underlying this reproducible and significant association of fusion structure and clinical behavior are unclear. As a first clue to how these variables may be linked, we have recently found that the EWS-FLI1 type 1 fusion may encode a transcription factor with lower transactivation efficiency than other EWS-FLI1 fusion types. 8 Because studies using various approaches to inhibit EWS-FLI1 function have shown that it is a critical determinant of proliferation in ES/PNET cell lines, 9-11 we have now sought to determine whether the observed covariation of structure, function, and clinical course extends to tumor cell kinetic parameters such as proliferative rate and apoptosis. We have also examined expression of the receptor for insulin-like growth factor 1 (IGF-1R), because there is evidence from several groups implicating it in autocrine or paracrine control of ES/PNET growth. 12-15 The results of our analysis, described below, are consistent with the notion that type 1 EWS-FLI1, functionally a weaker transcription factor, may result in lesser activation of direct or indirect target genes, possibly including IGF-1R, controlling proliferative rate in ES/PNET.
Materials and Methods
Patients
We studied 86 patients with a histopathological diagnosis of ES/PNET and molecular evidence of the EWS-FLI1 or EWS-ERG fusion transcripts, 66 from the Memorial Sloan-Kettering Cancer Center (MSKCC), 14 from Clínica Universitaria de Navarra (CUN), and 6 from the University of Pennsylvania Medical Center/Children’s Hospital of Philadelphia (UPMC/CHOP). Of 72 cases with the EWS-FLI1 fusion, 54 were included in our previous study. 7 The 14 EWS-ERG cases, consisting of 7 MSKCC cases, 6 UPMC/CHOP cases, and 1 case from CUN, were also included in another study. 16 EWS-ERG cases were actively sought and therefore their proportion in the overall series (16%) is not representative of their actual prevalence. There were 55 males and 31 females. The mean age was 23 years (range, 5–72 years). Primary tumor dimensions were available in 63 patients. Volume calculations for bony (spheroidal) and soft tissue (spherical) tumors were performed as described elsewhere. 17 Mean and median tumor volumes were, respectively, 456 ml and 72 ml.
Immunohistochemical and Apoptosis Analysis
All immunohistochemical and apoptosis analyses were done on 4-μm sections of formalin-fixed, paraffin-embedded, non-decalcified tumor. In 80 cases, prechemotherapy primary tumor was available for study, and in the remaining 6 cases metastases were used as the source of tumor. Immunohistochemistry was performed on an automated immunostainer (TechMate 500; Dako, Copenhagen, Denmark) with the EnVision+ system (Dako), in which the secondary antibody is coupled to a dextran polymer linked to peroxidase. Endogenous peroxidase activity was quenched by treatment with 5% hydrogen peroxide in methanol for 30 minutes at room temperature. Antigen retrieval by microwave treatment for 20 minutes in an 800W microwave oven was performed. A blocking step with normal rabbit serum was used. Primary antibodies were applied for 120 minutes at room temperature. Primary antibodies and dilutions were as follows: Ki-67 (MIB1 antibody, Zymed, South San Francisco, CA) 1/800 with microwave antigen retrieval, IGF-1R (1H7 antibody, RDI, Flanders, NJ) 1/1200 with microwave retrieval and trypsinization.
The sections were then rinsed with washing buffer at room temperature. The next step was addition of EnVision+ system reagents and incubation 30 minutes at room temperature. The slides were rinsed with washing buffer, and treated with a solution containing 0.05% diaminobenzidine hydrochloride and 0.1% hydrogen peroxide in 0.05 mol/L Tris-buffered saline, pH 7.4, at room temperature for 5 minutes. After rinsing in distilled water for 3 minutes, the slides were counterstained with modified Harris hematoxylin, dehydrated, and mounted. For negative controls, incubation with normal goat serum instead of primary antibody was used. Omission of the primary antibody likewise gave no background staining.
In situ detection of apoptosis by use of the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) technique was performed as previously described using the ApopTag In Situ Apoptosis Detection Kit (Intergen, Gaithersburg, MD) that identifies cells with internucleosomal fragmentation of DNA. Sections from selected formalin-fixed, paraffin-embedded tissue blocks were placed on coated slides. Briefly, tissue sections were dewaxed and rehydrated routinely. After rehydration, the slides were incubated with proteinase K (20 μg/ml) at room temperature for 15 minutes, per the manufacturer’s protocol. Endogenous peroxidase was inactivated by 3% hydrogen peroxide. Tissue sections were then subjected to the ApopTag reaction. The reaction was terminated after 60 minutes by transfer of the slides to a coplin jar containing pre-warmed working strength stop-wash buffer and incubated for 30 minutes at 37°C. The sections were then covered with 55 μl of anti-digoxigenin-peroxidase and incubated for 30 minutes at room temperature, and stained with diaminobenzidine to detect the labeled nuclei. For negative controls, deionized water was used instead of terminal deoxynucleotidyl transferase. Positive controls consisted of inflamed human tonsil and acute myocardial infarction. Cells were considered positive when brown reactivity was detected in the nuclei.
Results of immunohistochemical and apoptosis assays were evaluated by two independent observers (E. de A. and A. P.) blinded to clinicopathological and molecular data. Tissue sections were examined under high power magnification (×400). The number of immunoreactive cells was counted on three randomly selected fields, and expressed as a percentage of cells. In tumors with heterogeneous distribution of the immunoreactivity, four areas with the highest and the lowest number of reactive cells were counted and median value was reported. Cases in which discrepancy between observers was >15% were reviewed and a consensus was reached.
Molecular Study
The fusion transcripts in all but 18 cases had been characterized in previous studies. 7,16 Reverse transcriptase-polymerase chain reaction (RT-PCR) for EWS-FLI1 and EWS-ERG transcripts was performed on total RNA extracted from snap frozen tumor samples. EWS-FLI1 type 1 (7–6 fusion) and type 2 (7–5 fusion) products were typed according to the size of the RT-PCR products on agarose gel electrophoresis. Other less common fusion products were either blotted and probed with internal and junction-specific probes, or sequenced. EWS-ERG fusion transcripts were not subtyped for this analysis. Negative controls lacking either tumor RNA or reverse transcriptase were routinely used.
Statistical Analysis
Immunoreactivities with the different antibodies and apoptosis results were assessed as continuous variables. Because number of immunoreactive cells did not follow a normal distribution, we used Mann-Whitney nonparametric tests to compare qualitative (gene fusion type) with quantitative parameters (immunoreactivity). Logistic regression was used to study the univariate prognostic value of each factor for predicting proliferation, measured as Ki-67 index below or above the median value (15%). Factors with a univariate P value of not more than 0.2 for predicting Ki-67 index were considered in a multivariate logistic regression. The two factors included in final logistic regression, EWS-FLI1 transcript type, and IGF-1R expression were chosen to give the best two-factor model among all of the factors considered. All analyses were performed using SPSS statistical package, version 6.0 for Windows (SPSS, Inc. Chicago, IL). All P values were two-tailed.
Results
Immunoreactivity for Ki-67 obtained with the MIB1 antibody had a nuclear pattern with strong nucleolar staining (Figure 1) ▶ . Immunostaining for Ki-67 was most frequently seen in nuclei of dark cells, that seldom showed TUNEL reactivity. In ES/PNET, cells with hyperchromatic nuclei (dark cells) are usually seen along with cells having clear nuclei. They have traditionally been regarded as apoptotic cells. 18 Interestingly, nuclei reactive with the TUNEL technique were usually those of clear cells, whereas those of dark cells appeared positive less often. This pattern of TUNEL reactivity in some clear cells could represent an early stage of apoptosis, because the characteristic morphological appearance of apoptotic nuclei is a later event in the apoptotic process. Membranous staining was seen with the antibody to IGF-1R (Figure 2) ▶ . Most cases showed widespread IGF-1R immunoreactivity and all cases contained at least some positive cells.
Figure 1.

Ki-67 proliferation rate assesed by MIB1 immunohistochemistry in ES/PNET. Left panel shows ES/PNET case with non-type 1 EWS-FLI1 and high Ki-67. Right panel shows ES/PNET case with type 1 EWS-FLI1 and low Ki-67.
Figure 2.
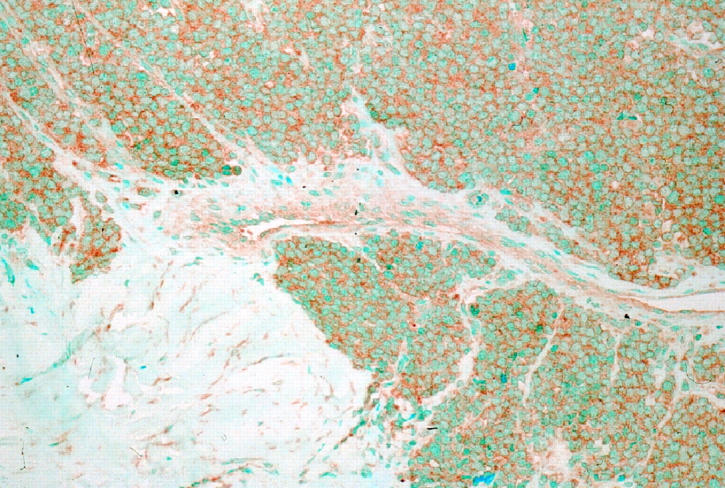
IGF-1R immunohistochemistry in ES/PNET. ES/PNET case with high IGF-1R expression is shown.
The mean and median percentages of tumor cells positive for these markers were respectively as follows: Ki-67 (n = 85) 21% and 14%, IGF-1R (n = 78) 73% and 80%, and TUNEL (n = 66) 5% and 2%. There was a significant positive correlation between proliferative index as assessed by immunostaining for Ki-67, and IGF-1R immunoreactivity (Spearman correlation coefficient, r = 0.23; P = 0.04). All cases with Ki-67 immunoreactivity in >20% of tumor cells had IGF-1R expression in 50% or more of cells (Figure 3) ▶ . Conversely, all cases with <40% IGF-1R-positive cells had a Ki-67 proliferative index of 10% or less.
Figure 3.

Scattergram showing correlation between Ki-67 (MIB1) and IGF-1R immunoreactivities. Significant Ki-67 immunoreactivity (>10%) was only present when over 50% of cells show expression of IGF-1R. Tumors having non-type 1 EWS-FLI1 fusions (green triangles) had significantly higher values of IGF-1R and Ki-67 (see Tables 1 and 2 ▶ ▶ ).
There were 72 tumors with EWS-FLI1 transcripts: 44 type 1 fusions and 28 grouped into the non-type 1 group. Fourteen tumors had EWS-ERG fusions. Patients having EWS-FLI1 fusions had similar age, stage at diagnosis, and tumor volume with respect to those bearing EWS-ERG fusion genes (results not shown).
In the group of tumors with EWS-FLI1 transcripts, those having type 1 fusions had a lower immunoreactivity for Ki-67 (15%; P = 0.049), and for IGF-1R (65%; P = 0.015) than tumors with other EWS-FLI1 fusion types (24% and 82%, respectively) by Mann-Whitney test (Table 1) ▶ . In aggregate, tumors with EWS-FLI1 transcripts had lower values for Ki-67 (P = 0.01) than those with EWS-ERG fusions by Mann-Whitney test (Table 1) ▶ .
Table 1.
Comparison of TUNEL, Ki-67, and IGF-1R Results as Continuous Variables
| Parameter | EWS-FLI1 type 1 | EWS-FLI1 non-type 1 | P value* | EWS-FLI1 (all types) | EWS-ERG | P value* |
|---|---|---|---|---|---|---|
| TUNEL | 2.4 ± 0.4 | 5.3 ± 3.6 | 0.49 | 3.5 ± 1.4 | 10.1 ± 5.9 | 0.3 |
| (n = 66) | ||||||
| Ki-67 | 15 ± 2 | 24 ± 4 | 0.049 | 18 ± 2 | 35 ± 8 | 0.01 |
| (n = 85) | median, 12 | median, 20 | median, 13 | median, 23 | ||
| IGF-1R | 65 ± 5 | 82 ± 4 | 0.015 | 72 ± 3 | 79 ± 5 | 0.58 |
| (n = 78) | median, 73 | median, 85 | median, 80 | median, 80 |
*Mann-Whitney test.
Values correspond to mean percentage value of immunoreactive cells ± SE.
We then analyzed Ki-67 proliferation index as a categorical variable by using 15%, essentially the median Ki-67 value in the entire study group (see above), to delineate low and high proliferation subgroups. By Fisher’s exact test, both the type 1 versus non-type 1 EWS-FLI1 comparison (P = 0.047; Table 2 ▶ ) and the EWS-FLI1 versus EWS-ERG comparison (P = 0.02; Table 3 ▶ ) remained statistically significant.
Table 2.
Comparison of Ki-67 Results as Categorical Variables, EWS-FLI1 Type 1 vs. EWS-FLI1 Non-Type 1
| Category | EWS-FLI1 type 1 | EWS-FLI1 non-type 1 |
|---|---|---|
| Ki-67 < 15% | 30 | 11 |
| Ki-67 > 15% | 15 | 16 |
P = 0.047, Fisher’s exact test.
Values correspond to number of cases.
Table 3.
Comparison of Ki-67 Results as Categorical Variables, EWS-FLI1 vs. EWS-ERG
| Category | EWS-FLI1* | EWS-ERG* |
|---|---|---|
| Ki-67 < 15% | 41 | 3 |
| Ki-67 > 15% | 31 | 11 |
P = 0.02, Fisher’s exact test.
Values correspond to number of cases.
*all types.
To further dissect the relative contributions of these variables to proliferation index in ES/PNET, we performed a logistic regression analysis of factors determining Ki-67. In an analysis including EWS-FLI1 type 1 versus non-type 1 and IGF-1R, the latter remained significant (P = 0.049), but EWS-FLI1 transcript type was no longer significant (P = 0.3), suggesting that the impact of the latter on Ki-67 values may be due largely to its correlation with IGF-1R expression.
No significant correlations were seen between Ki-67 or transcript type and stage at diagnosis or tumor volume. Tumors metastatic at diagnosis were larger than those presenting with localized disease only (1390 ml vs. 242 ml respectively; P = 0.001). No other correlations were identified among the parameters studied. There were no significant differences in the proportion of apoptotic cells, as detected by the TUNEL assay, among the groups.
Discussion
The consistent involvement of transcription factors in the gene fusions observed in many small cell sarcomas suggests a central role for these proteins in the regulation of proliferation and differentiation of mesenchymal cells. 19 EWS-FLI1 is an aberrant transcription factor, and experimental evidence suggests that EWS-FLI1 initiates and maintains tumorigenicity in ES/PNET. 2 Indeed, specific inactivation of EWS-FLI1 in ES/PNET cell lines results in reduced proliferation and loss of tumorigenicity. 9-11
Ki-67 is expressed in late G1, S, G2, and M phases, and hence correlates closely with growth fraction in all tumor types. 20 Although its function is not well known, nuclear overexpression of Ki-67 has been associated with tumor progression and poor prognosis in many different tumors, including sarcomas. 21-24 There have been few systematic studies of proliferation markers in ES/PNET. Our median Ki-67 value with the MIB1 antibody (14%) was slightly higher than the range of proliferative rates previously reported for this tumor (7–10%) using other techniques. 25-28 The significant positive correlation between proliferative rate and IGF-1R expression level (P = 0.04) found in the present study is consistent with the known cellular biology of IGF-1R 29,30 and supports the validity of the immunohistochemical methods used to evaluate these two parameters.
Our Ki-67 data, whether analyzed as continuous or categorical variables, suggest that tumors with EWS-FLI1 type 1 have, on average, a lower proliferative rate than ES/PNET with non-type 1 EWS-FLI1. Our logistic regression analysis further suggests that the partial association of type 1 EWS-FLI1 and lower IGF-1R expression may account at least in part for the association of Ki-67 and EWS-FLI1 fusion type.
IGF-1R is widely expressed by proliferating cells throughout development. 29,30 IGF-1 and its receptor, IGF-1R, are expressed in many human tumors, 29,30 including various sarcomas. 31 In at least some ES/PNET, they appear to constitute a functional autocrine or paracrine loop. 12,14 Blockade of IGF-1R stops proliferation and induces apoptosis in ES/PNET. 12-14 Consistent with these studies, we found a significant positive correlation in ES/PNET between Ki-67 proliferative index and the proportion of cells expressing IGF-1R. Tumors with EWS-FLI1 Type 1 fusions had significantly fewer cells immunoreactive for IGF-1R. The simplest model accounting for these associations of fusion type, IGF-1R expression, and proliferative rate would suggest that the IGF-1R gene is a direct or indirect target of EWS-FLI1 and therefore subject to differential regulation by alternative forms of this aberrant transcription factor. 8 Few regulatory targets of EWS-FLI1 or native FLI1 have so far been identified. 32-35 There are contradictory data on IGF-1R regulation by EWS-FLI1. In a study recently reported in abstract form, Shum et al 36 found IGF-1R gene expression to be decreased following induction of EWS-FLI1 in a human embryonal kidney cell line (EcR293). On the other hand, Toretsky et al reported no significant difference in endogenous IGF-1R protein levels in mouse fibroblast cell lines with or without stably transfected human EWS-FLI1, 37 leading them to conclude that IGF-1R expression is not regulated by the fusion protein. Whether these experimental models are representative of IGF-1R regulation in ES/PNET is unclear, however. The human and murine IGF-1R promoter regions are not identical, and mouse fibroblasts, human embryonal kidney cells, and human ES/PNET cells may differ in cell type-specific transcriptional cofactors.
We found all cases to show at least some IGF-1R expression by immunohistochemistry. This is consistent with its ubiquity at the mRNA or protein level in ES/PNET cell lines or clinical samples as shown by others. 12,14 Evidence that IGF-1R expression is required for EWS-FLI1 transformation of mouse fibroblasts 37 suggests that in vivo, this fusion protein may have a similar requirement for a IGF-1R-positive precursor cell or for induction of the IGF-1R pathway. Although essentially all cases showed at least some IGF-1R expression, its level could be critical for maintaining cell proliferation in ES/PNET, because only cases with >40% of cells expressing IGF-1R had a significant Ki-67 immunoreactivity (>10%; see Figure 3 ▶ ). The differences in extent of IGF-1R immunoreactivity between type 1 and non-type 1 EWS-FLI1 groups, although statistically significant (P = 0.015), were relatively small (65% vs. 82%). Nonetheless, there is experimental evidence that small differences in IGF-1R expression level per cell can effectively modulate cell proliferation. 38
We have recently reported in a partially overlapping series that tumors with EWS-ERG fusions have a similar clinical presentation and behavior as those with EWS-FLI1 chimeric genes. 16 It may thus be unexpected that there was a difference in proliferative index, as assessed by Ki-67 immunoreactivity, between these two groups. No differences in IGF-1R expression were seen between EWS-FLI1 and EWS-ERG cases, suggesting that the observed difference in proliferative rate could be due to differential activation of other target pathways. The extent to which activation of downstream targets may differ quantitatively or qualitatively between EWS-FLI1 and EWS-ERG is unknown. Indeed, outside their DNA-binding domains, FLI1 and ERG show major sequence differences which could be significant in the protein-protein interactions involved in the regulation of certain promoters. It is therefore not unreasonable to consider the possibility that EWS-FLI1 and EWS-ERG may deregulate some partly non-overlapping pathways to generate clinical phenotypes which are so far indistinguishable.
Because both EWS-FLI1 and EWS-ERG fusion proteins have been shown to inhibit apoptosis in model systems, 39 we were interested in comparing relative numbers of apoptotic cells in these pretreatment clinical samples. However, the numbers of apoptotic cells shown by the TUNEL technique were small and there were no significant differences among the fusion types.
Other determinants of proliferation rate may include secondary genetic alterations in cell cycle regulators. In two series of ES/PNET which partially overlapped with the present study group, we have found evidence of P53 alterations in 11% and INK4A deletion in 17%. 40,41 Similar percentages have been reported by other groups. 42-48 These genetic alterations are relatively uncommon but appear prognostically significant. 40,41,48 Because of their low prevalence, however, alterations in cell cycle regulators are unlikely to account for the significant differences in Ki-67 values between groups in the present study, but may explain some of the variability in proliferative rate within these groups. Establishing whether the distribution of these secondary genetic alterations in cell cycle regulators is random in relation to EWS-ETS fusion type will require a much larger systematic multiparameter study.
In summary, the most common type of EWS-FLI1 fusion transcript, type 1, is associated with a favorable prognosis 6,7 and appears to encode a functionally weaker transactivator 8 compared to other fusion types. The present study indicates that these differences are paralleled by similar differences in proliferative rate, perhaps mediated in part by the putative differential regulation of the IGF-1R pathway by these alternative forms of EWS-FLI1. Further elucidation of the target genes of EWS-FLI1 should clarify this intriguing aspect of the biology of ES/PNET.
Acknowledgments
We thank Denis Leung, Ph.D., for statistical advice and review, Margaret Collins, M.D., for providing paraffin sections of blocks from the Department of Pathology at the Children’s Hospital of Philadelphia and Loli Martinez for technical assistance with immunohistochemical and TUNEL assays.
Footnotes
Address reprint requests to Marc Ladanyi, M.D., Department of Pathology, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10021. E-mail: ladanyim@mskcc.org.
Supported in part by American Cancer Society Research Project Grant 99–216 (to M. L.) and by the Public Health Care System of Spain, Madrid, Spain (FIS grant 96/2102 to E. de A).
References
- 1.Delattre O, Zucman J, Melot T, Garau XS, Zucker JM, Lenoir GM, Ambros PF, Sheer D, Turc-Carel C, Triche TJ, Aurias A, Thomas G: The Ewing family of tumors: a subgroup of small-round-cell tumors defined by specific chimeric transcripts. New Engl J Med 1994, 331:294-299 [DOI] [PubMed] [Google Scholar]
- 2.Kovar H: Progress in the molecular biology of Ewing tumors. Sarcoma 1998, 2:3-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barr FG, Chatten J, D’Cruz CM, Wilson AE, Nauta LE, Nycum LM, Biegel JA, Womer RB: Molecular assays for chromosomal translocations in the diagnosis of pediatric soft tissue sarcomas. J Am Med Assoc 1995, 273:553-557 [PubMed] [Google Scholar]
- 4.de Alava E, Ladanyi M, Rosai J, Gerald WL: Detection of chimeric transcripts in desmoplastic small round cell tumor and related developmental tumors by reverse transcriptase polymerase chain reaction: a specific diagnostic assay. Am J Pathol 1995, 147:1584-1591 [PMC free article] [PubMed] [Google Scholar]
- 5.Zucman J, Melot T, Desmaze C, Ghysdael J, Plougastel B, Peter M, Zucker JM, Triche TJ, Sheer D, Turc-Carel C, Ambros P, Combaret V, Lenoir G, Aurias A, Thomas G, Delattre O: Combinatorial generation of variable fusion proteins in the Ewing family of tumours. EMBO J 1993, 12:4481-4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zoubek A, Dockhorn-Dworniczak B, Delattre O, Christiansen H, Niggli F, Gatterer-Menz I, Smith TL, Jurgens H, Gadner H, Kovar H: Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J Clin Oncol 1996, 14:1245-1251 [DOI] [PubMed] [Google Scholar]
- 7.de Alava E, Kawai A, Healey JH, Fligman I, Meyers P, Huvos AG, Gerald WL, Jhanwar SC, Argani P, Antonescu CR, Pardo-Mindan FJ, Ginsberg J, Womer R, Lawlor ER, Wunder J, Andrulis I, Sorensen PHB, Barr FG, Ladanyi M: EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. J Clin Oncol 1998, 16:1248-1255 [DOI] [PubMed] [Google Scholar]
- 8.Lin PP, Brody RI, Hamelin A, Bradner JE, Healey JH, Ladanyi M: Differential transactivation by alternative EWS-FLI1 fusion proteins correlates with clinical heterogeneity in Ewing’s sarcoma. Cancer Res 1999, 59:1428-1432 [PubMed] [Google Scholar]
- 9.Kovar H, Aryee DN, Jug G, Henockl C, Schemper M, Delattre O, Thomas G, Gadner H: EWS/FLI-1 antagonists induce growth inhibition of Ewing tumor cells in vitro. Cell Growth Differ 1996, 7:429-437 [PubMed] [Google Scholar]
- 10.Ouchida M, Ohno T, Fujimura Y, Rao VN, Reddy ESP: Loss of tumorigenicity of Ewing’s sarcoma cells expressing antisense RNA to EWS-fusion transcripts. Oncogene 1995, 11:1049-1054 [PubMed] [Google Scholar]
- 11.Tanaka K, Iwakuma T, Harimaya K, Sato H, Iwamoto Y: EWS-FLI1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing’s sarcoma, and primitive neuroectodermal tumor cells. J Clin Invest 1997, 99:239-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yee D, Favoni RE, Lebovic GS, Lombana F, Powell DR, Reynolds CP, Rosen N: Insulin-like growth factor I expression by tumors of neuroectodermal origin with the t(11;22) chromosomal translocation: a potential autocrine growth factor. J Clin Invest 1990, 86:1806-1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chin LS, Yung WK, Raffel C: Two primitive neuroectodermal tumor cell lines require an activated insulin-like growth factor I receptor for growth in vitro. Neurosurgery 1996, 39:1183-1190 [DOI] [PubMed] [Google Scholar]
- 14.Scotlandi K, Benini S, Sarti M, Serra M, Lollini PL, Maurici D, Picci P, Manara MC, Baldini N: Insulin-like growth factor I receptor-mediated circuit in Ewing’s sarcoma/peripheral neuroectodermal tumor: a possible therapeutic target. Cancer Res 1996, 56:4570-4574 [PubMed] [Google Scholar]
- 15.Scotlandi K, Benini S, Nanni P, Lollini P-L, Nicoletti G, Landuzzi L, Serra M, Manara MC, Picci P, Baldini N: Blockage of insulin-like growth factor-I receptor inhibits the growth of Ewing’s sarcoma in athymic mice. Cancer Res 1998, 58:4127-4131 [PubMed] [Google Scholar]
- 16.Ginsberg JP, de Alava E, Ladanyi M, Wexler L, Kovar H, Paulussen M, Zoubek A, Dockhorn-Dworniczak BH, Juergens H, Wunder JS, Andrulis IL, Malik R, Sorensen PHB, Womer RB, Barr FG: EWS-FLI1, and EWS-ERG gene fusions are associated with similar clinical phenotypes in Ewing’s sarcoma. J Clin Oncol 1999, 17:1809-1814 [DOI] [PubMed] [Google Scholar]
- 17.Picci P, Bohling T, Bacci G, Ferrari S, Sangiorgi L, Mercuri M, Ruggieri P, Manfrini M, Ferraro A, Casadei R, Benassi MS, Mancini AF, Rosito P, Cazzola A, Barbieri E, Tienghi A, Brach del Prever A, Comandone A, Bacchini P, Bertoni F: Chemotherapy-induced tumor necrosis as a prognostic factor in localized Ewing’s sarcoma of the extremities. J Clin Oncol 1997, 15:1553–1559 [DOI] [PubMed]
- 18.Terrier P, Llombart-Bosch A, Contesso G: Small round blue cell tumors in bone: prognostic factors correlated to Ewing’s sarcoma and neuroectodermal tumors. Semin Diagn Pathol 1996, 13:250-257 [PubMed] [Google Scholar]
- 19.Barr FG: Translocations, cancer and the puzzle of specificity. Nat Genet 1998, 19:121-124 [DOI] [PubMed] [Google Scholar]
- 20.Levine EA, Holzmayer T, Bacus S, Mechetner E, Mera R, Bolliger C, Roninson IB, Das GT: Evaluation of newer prognostic markers for adult soft tissue sarcomas. J Clin Oncol 1997, 15:3249–3257 [DOI] [PubMed]
- 21.Choong PF, Akerman M, Willen H, Andersson C, Gustafson P, Baldetorp B, Ferno M, Alvegard T, Rydholm A: Prognostic value of Ki-67 expression in 182 soft tissue sarcomas. Proliferation: a marker of metastasis? APMIS 1994, 102:915-924 [DOI] [PubMed] [Google Scholar]
- 22.Drobnjak M, Latres E, Pollack D, Karpeh M, Dudas M, Woodruff JM, Brennan MF, Cordon-Cardo C: Prognostic implications of p53 nuclear overexpression and high proliferation index of Ki-67 in adult soft-tissue sarcomas. J Natl Cancer Inst 1994, 86:549-554 [DOI] [PubMed] [Google Scholar]
- 23.Heslin MJ, Cordon-Cardo C, Lewis JJ, Woodruff J, Brennan MF: Ki-67 detected by MIB-1 predicts distant metastasis and tumor mortality in primary, high grade extremity soft tissue sarcoma. Cancer 1998, 83:490-497 [DOI] [PubMed] [Google Scholar]
- 24.Jensen V, Sorensen FB, Bentzen SM, Ladekarl M, Nielsen OS, Keller J, Jensen OM: Proliferative activity (MIB-1 index) is an independent prognostic parameter in patients with high-grade soft tissue sarcomas of subtypes other than malignant fibrous histiocytomas: a retrospective immunohistological study including 216 soft tissue sarcomas. Histopathology 1998, 32:536-546 [PubMed] [Google Scholar]
- 25.Vollmer E, Roessner A, Wuisman P, Harle A, Grundmann E: The proliferation behavior of bone tumors investigated with the monoclonal antibody Ki-67. Curr Top Pathol 1989, 80:91-114 [DOI] [PubMed] [Google Scholar]
- 26.Mellin W, Dierschauer W, Hiddemann W, Roessner A, Edel G, Wuisman P, Harle A, Grundmann E: Flow cytometric DNA analysis of bone tumors. Curr Top Pathol 1989, 80:115-152 [DOI] [PubMed] [Google Scholar]
- 27.Niggli FK, Powell JE, Parkes SE, Ward K, Raafat F, Mann JR, Stevens MC: DNA ploidy and proliferative activity (S-phase) in childhood soft-tissue sarcomas: their value as prognostic indicators. Br J Cancer 1994, 69:1106-1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scotlandi K, Serra M, Manara MC, Maurici D, Benini S, Nini G, Campanacci M, Baldini N: Clinical relevance of Ki-67 expression in bone tumors. Cancer 1995, 75:806-814 [DOI] [PubMed] [Google Scholar]
- 29.Baserga R, Resnicoff M, Dews M: The IGF-I receptor, and cancer. Endocrine 1997, 7:99-102 [DOI] [PubMed] [Google Scholar]
- 30.Blakesley VA, Stannard BS, Kalebic T, Helman LJ, Le Roith D: Role of the IGF-I receptor in mutagenesis and tumor promotion. J Endocrinol 1997, 152:339-344 [DOI] [PubMed] [Google Scholar]
- 31.Sekyi-Otu A, Bell RS, Ohashi C, Pollak M, Andrulis IL: Insulin-like growth factor 1 (IGF-1) receptors, IGF-1, and IGF-2, are expressed in primary human sarcomas. Cancer Res 1995, 55:129-134 [PubMed] [Google Scholar]
- 32.Braun BS, Frieden R, Lessnick SL, May WA, Denny CT: Identification of target genes for the Ewing’s sarcoma EWS/FLI fusion protein by representational difference analysis. Mol Cell Biol 1995, 15:4623-4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.May WA, Arvand A, Thompson AD, Braun BS, Wright M, Denny CT: EWS/FLI1-induced manic fringe renders NIH 3T3 cells tumorigenic. Nat Genet 1997, 17:495-497 [DOI] [PubMed] [Google Scholar]
- 34.Arvand A, Bastians H, Welford SM, Thompson AD, Ruderman JV, Denny CT: EWS/FLI1 up-regulates mE2-C, a cyclin-selective ubiquitin conjugating enzyme involved in cyclin B destruction. Oncogene 1998, 17:2039-2045 [DOI] [PubMed] [Google Scholar]
- 35.Bastian LS, Kwiatkowski BA, Breininger J, Danner S, Roth G: Regulation of the megakaryocytic glyucoprotein IX promoter by the oncogenic Ets transcription factor Fli-1. Blood 1999, 93:2637-2644 [PubMed] [Google Scholar]
- 36.Shum CH, Jackson G, Haynes JA, Liu XF, Triche TJ: Downstream targets of the Ewing’s sarcoma EWS/FLI-1 fusion gene. Proc Am Assoc Cancer Res 1999, 40:321(abstract) [Google Scholar]
- 37.Toretsky JA, Kalebic T, Blakesley V, LeRoith D, Helman LJ: The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J Biol Chem 1997, 272:30822-30827 [DOI] [PubMed] [Google Scholar]
- 38.Rubini M, Hongo A, D’Ambrosio C, Baserga R: The IGF-I receptor in mitogenesis, and transformation of mouse embryo cells: role of receptor number. Exp Cell Res 1997, 230:284-292 [DOI] [PubMed] [Google Scholar]
- 39.Yi H, Fujimura Y, Ouchida M, Prasad DD, Rao VN, Reddy ES: Inhibition of apoptosis by normal and aberrant Fli-1 and erg proteins involved in human solid tumors and leukemias. Oncogene 1997, 14:1259-1268 [DOI] [PubMed] [Google Scholar]
- 40.de Alava E, Antonescu CR, Panizo A, Leung DH, Meyers P, Huvos AG, Pardo-Mindan FJ, Healey JH, Ladanyi M: Secondary genetic alterations in Ewing’s sarcoma: prognostic impact of P53 status. Cancer (in press) [PubMed]
- 41.Wei G, Antonescu CR, de Alava E, Leung D, Huvos AG, Meyers P, Healey JH, Ladanyi M: Seconary genetic alterations in Ewing’s sarcoma: prognostic impact of INK4A deletion. Cancer (in press) [DOI] [PubMed]
- 42.Kovar H, Auinger A, Jug G, Aryee D, Zoubek A, Salzer-Kuntschik M, Gadner H: Narrow spectrum of infrequent p53 mutations and absence of MDM2 amplification in Ewing tumours. Oncogene 1993, 8:2683-2690 [PubMed] [Google Scholar]
- 43.Komuro H, Hayashi Y, Kawamura M, Hayashi K, Kaneko Y, Kamoshita S, Hanada R, Yamamoto K, Hongo T, Yamada M, Tsuchida Y: Mutations of the p53 gene are involved in Ewing’s sarcomas but not in neuroblastomas. Cancer Res 1993, 53:5284-5288 [PubMed] [Google Scholar]
- 44.Hamelin R, Zucman J, Melot T, Delattre O, Thomas G: P53 mutations in human tumors with chimeric EWS/FLI-1 genes. Int J Cancer 1994, 57:336-340 [DOI] [PubMed] [Google Scholar]
- 45.Mangham D, Cannon A, Li X, Komiya S, Gebhardt M, Springfield D, Rosenberg A, Mankin HJ: p53 overexpression in Ewing’s sarcoma/primitive neuroectodermal tumour is an uncommon event. J Clin Pathol Mol Pathol 1995, 48:M79-M82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mousses S, McAuley L, Bell RS, Kandel R, Andrulis IL: Molecular and immunohistochemical identification of p53 alterations in bone and soft tissue sarcomas. Mod Pathol 1996, 9:1-6 [PubMed] [Google Scholar]
- 47.Kovar H, Jug G, Aryee DNT, Zoubek A, Ambros P, Gruber B, Windhager R, Gadner H: Among genes involved in the RB dependent cell cycle regulatory cascade, the p16 tumor suppressor gene is frequently lost in the Ewing family of tumors. Oncogene 1997, 15:2225-2232 [DOI] [PubMed] [Google Scholar]
- 48.Abudu A, Mangham DC, Reynolds GM, Pynsent PB, Tillman RM, Carter SR, Grimer RJ: Overexpression of p53 protein in primary Ewing’s sarcoma of bone: relationship to tumour stage, response and prognosis. Br J Cancer 1999, 79:1185-1189 [DOI] [PMC free article] [PubMed] [Google Scholar]