

Abstract
Patients harboring germline BRCA2 mutations are at an increased risk of developing pancreatic cancer. We investigated the prevalence of biallelic inactivation of BRCA2 in the presumed precursors to invasive pancreatic ductal carcinomas, pancreatic intraepithelial neoplasia (PanIN). Surgical resection specimens from three patients with germline BRCA2 mutations who developed pancreatic ductal adenocarcinoma were studied. Fourteen PanINs were needle-microdissected from paraffin-embedded tissue. DNA was isolated from these microdissected tissues and amplified by primer-mediated pre-amplification. Loss of heterozygosity at the BRCA2 locus was determined by polymerase chain reaction amplification and cycle sequencing. The presence of the wild-type alleles was evaluated at the nucleotide positions of the germline BRCA2 mutations. The K-ras gene was sequenced at codon 12 and 13 to confirm the efficacy of microdissection. By histological evaluation the prevalence of PanINs in these patients was not notably elevated. Loss of the wild-type allele of BRCA2 was present in one high-grade PanIN (PanIN 3), but in none of 13 low-grade PanINs (PanIN 1). In contrast, K-ras mutations were detectable in 7 of the 14 PanINs. These results suggest that biallelic inactivation of the BRCA2 gene is a relatively late event in pancreatic tumorigenesis. In contrast to classical molecular progression models of tumorigenesis, the inactivation of the wild-type allele in a carrier of a recessive tumor susceptibility gene may not always be the first somatic event during the molecular evolution of a cancer. The necessity for earlier genetic alterations before biallelic inactivation of a recessive tumor susceptibility gene such as BRCA2 may explain why affected carriers have normal numbers of neoplastic precursor lesions, a relatively low phenotypic penetrance, and late age of onset of pancreatic and other cancers.
Hereditary predisposition to adenocarcinoma of the pancreas is clinically evident in approximately 10% of patients who develop the disease. 1-3 Germline BRCA2 mutations account for a portion of this group, and as many as 5 to 10% of patients with apparently sporadic pancreatic cancer harbor germline BRCA2 mutations. 4,5 The lifetime risk of pancreatic cancer in carriers of a BRCA2 germline mutation is, however, probably in the range of 5%. 4-8 The genetic and environmental influences that result in the low penetrance of pancreatic cancer in BRCA2 mutant carriers are poorly understood. A critical genetic event that would be expected to influence penetrance of pancreatic cancer in these carriers is the occurrence and timing of inactivation of the wild-type BRCA2 allele in pancreatic epithelium. For example, in patients with familial adenomatous polyposis (FAP), the inactivation of the wild-type allele of APC in colonic epithelial stem cells is probably the first genetic alteration (the gatekeeper) during the evolution of a neoplasm. Loss of the wild-type APC allele occurs in early adenomas both in FAP patients and the MIN mouse. 9-12 In breast and pancreatic cancers that occur in carriers with germline BRCA2 mutations, the inactivation of the wild-type allele is usual. 4,13 Yet the timing of loss of the wild-type copy of BRCA2 has not been studied in neoplastic precursor lesions that develop in patients with germline BRCA2 mutations, and there are reasons to suspect that BRCA2 does not follow the gatekeeper model.
To study the timing of BRCA2 alterations in the development of pancreatic cancer we took advantage of the observation that multiple neoplastic precursor lesions are often present in the pancreata of patients with pancreatic ductal adenocarcinomas. 14-20 These lesions are called pancreatic intraepithelial neoplasia (PanIN; see http://www.path.jhu.edu/pancreas_panin). By determining the frequency of genetic alterations in the PanINs of varying histological severity, one can establish a progression model for the development of infiltrating adenocarcinoma of the pancreas. The histological and genetic analysis of such neoplastic precursors can provide insights into the genetic progression of pancreatic cancer in individuals with germline BRCA2 mutations.
We investigated the timing of biallelic inactivation of the BRCA2 gene in PanINs by analyzing DNA from a series of microdissected PanINs located in the pancreatic parenchyma adjacent to invasive pancreatic carcinomas resected from patients with germline BRCA2 mutations. DNA from these PanINs was analyzed for loss of the wild-type allele at BRCA2 and for the presence of K-ras gene mutations.
Materials and Methods
Patients with germline BRCA2 mutations who developed pancreatic cancers were identified previously. 4 From the archives of The Johns Hopkins Hospital, slides of pancreatic resection specimens from these cases with germline BRCA2 mutations were reviewed. Three cases were selected based on the presence of infiltrating pancreatic adenocarcinoma of the pancreas and the availability of archival material adequate for the study of associated PanINs (Table 1) ▶ . PanINs were classified using criteria established at the National Cancer Institute-sponsored Pancreatic Cancer Think Tank in Park City, Utah (http://www.path.jhu.edu/pancreas_panin). Briefly, PanINs were classified as grade 1 when duct lesions lacked significant nuclear abnormalities, as grade 2 when duct lesions had moderate cytological and architectural atypia, and grade 3 when those lesions showed marked architectural or cytologic atypia. The histological features were graded by an experienced pathologist (R. H. H.) familiar with the Park City classification scheme before the molecular analysis.
Table 1.
BRCA2 Allelic Loss and K-ras Gene Mutation in Pancreatic Intraepithelial Neoplasia (PanIN)
| Patient | PanIN grade | LOH at BRCA2 | K-ras codon 12 |
|---|---|---|---|
| PX101 | 3 | Yes | GAT |
| 1 | No | GAT | |
| 1 | No | GAT | |
| 1 | No | GGT | |
| 1 | No | GGT | |
| 1 | No | GGT | |
| 1 | No | GGT | |
| PX66 | 1 | No | GAT |
| 1 | No | GGT | |
| 1 | No | GAT | |
| 1 | No | Not determined | |
| PX182 | 1 | No | GTT |
| 1 | No | GGT | |
| 1 | No | GAT |
The germline mutations of BRCA2 in cases PX101, PX66, and PX182 were 2481insT, 6174delT, and 6158insT, respectively. PX101 and PX66 had LOH of BRCA2 identified in the carcinoma samples.
Seven-micron sections from formalin-fixed, paraffin-embedded tissue blocks were stained with hematoxylin and eosin. PanINs, infiltrating carcinoma, and adjacent normal tissue were dissected under direct visualization using an inverted microscope and a glass needle attached to a micromanipulator. 21 Mock dissections were performed on slides lacking tissue, from which the tip of the glass needle was processed for DNA isolation and polymerase chain reaction (PCR) amplification to examine for and rule out contamination during the process of microdissection. DNA was extracted from microdissected tissue using 500 μg/ml of proteinase K and 0.5% NP40 and incubated overnight at 56°C. DNA samples were then subjected to whole genome amplification by primer-mediated pre-amplification (PEP) as previously described. 22 We have previously observed stochastic errors in PCR when amplifying paraffin-embedded DNA when the input DNA is less than ∼50 cells, particularly if the PCR products are over 400 bp. 21 Hence, PEP was performed on 300 to 600 microdissected cells and amplified using a degenerate 15-mer for 50 cycles of 92° for 30 seconds, 37° for 2 minutes, and 55° for 4 minutes, ramping at 1° every 10 seconds. The reproducibility of PEP to amplify two alleles equally was determined by PEP amplification of DNA isolated from normal pancreatic acini and subsequently amplifying with PCR primers that specifically amplified the region of DNA spanning the patients’ known germline BRCA2 mutations. These results obtained using PEP were also confirmed by direct PCR amplification and sequencing of microdissected tissues in the absence of PEP. Sequencing of PCR products confirmed that PEP had amplified both BRCA2 alleles from all 18 samples. PCR was also performed using primers to amplify across the region of the K-ras gene containing codons 12 and 13. PCR products were subsequently analyzed by DNA cycle sequencing as described. 23 Loss of heterozygosity (LOH) at the BRCA2 locus was determined by the absence of the wild-type nucleotide sequence at the site of the germline mutation.
Results
The number and morphology of PanIN lesions were reviewed by obtaining all available hematoxylin-and-eosin-stained archival slides of the pancreatic carcinoma resection specimens from the three patients with known germline BRCA2 mutations. All three patients had undergone a Whipple resection. There were no observable qualitative or quantitative differences in the number or morphology of the PanINs compared to that seen in patients without germline BRCA2 mutations.
Three PanINs were selected from case PX182, four PanIN from case PX66, and seven PanIN from case PX101 for microdissection. The PX series comprises unique patients whose carcinomas were expanded by xenografting to allow genetic analysis. These 14 PanINs included 13 PanIN-1 and one PanIN-3. The germline mutations in the three cases were 6158insT, 6174delT, and 2481insT, respectively, as previously described. 4 Loss of the wild-type allele was evident in xenografts of the pancreatic adenocarcinoma in two of the three cases (6174delT and 2481insT). 4 LOH at the BRCA2 gene locus was present in the single PanIN-3 (from PX101), but in none of 13 low grade duct lesions (PanIN-1; Figures 1 and 2 ▶ ▶ ). LOH was not detected in three normal ducts, nor was it detected in multiple microdissections of pancreatic acini containing ∼1000–2000 cells.
Figure 1.
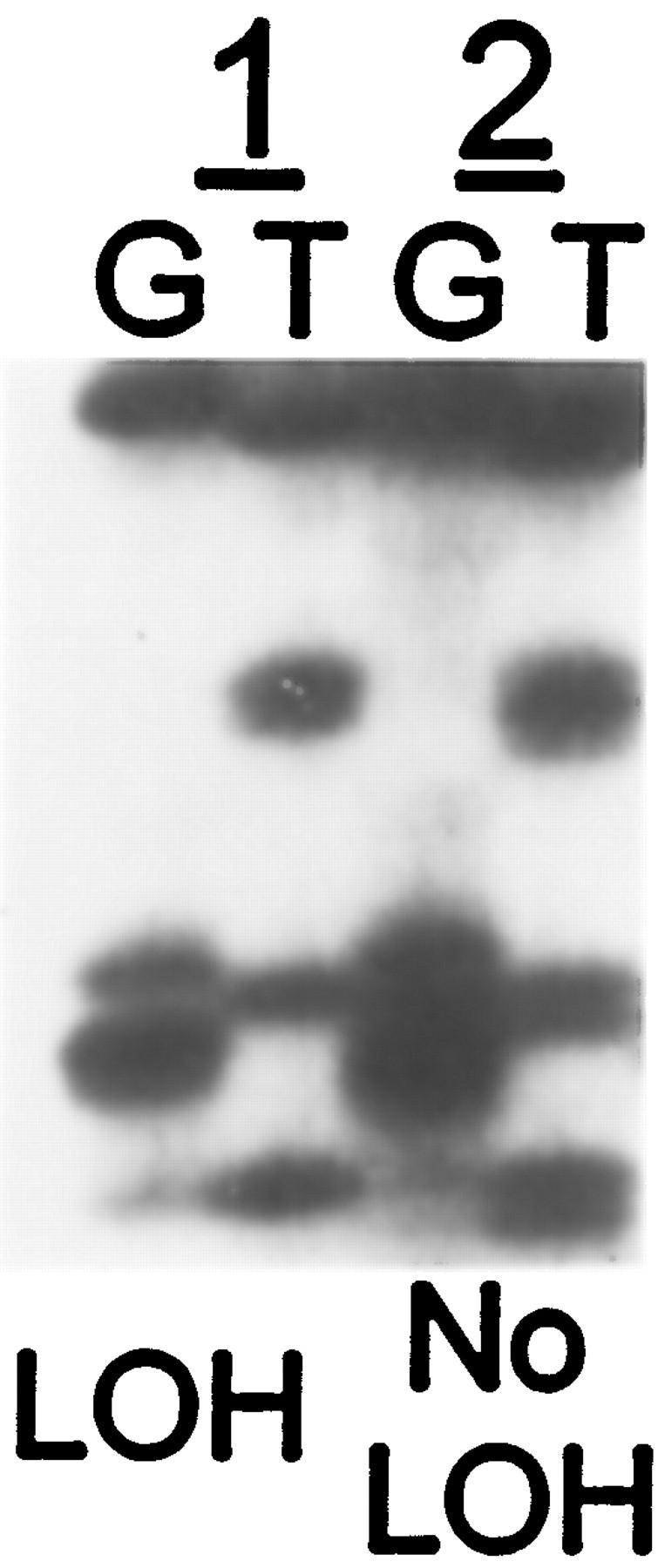
Sequence of the BRCA2 gene upstream of the 2481insT germline mutation in two PanIN lesions from a patient with pancreas cancer (case PX101). The first duct lesion (lane pair 1, PanIN-3) shows the mutant sequence with loss of the wild-type BRCA2 sequence, whereas the second lesion (lane pair 2, PanIN-1) shows the sequence of both the wild-type and mutant alleles (note the double bands for each nucleotide, best seen on comparison of the T lanes). G and T refer to deoxyguanosine and deoxythymidine termination reactions, respectively.
Figure 2.
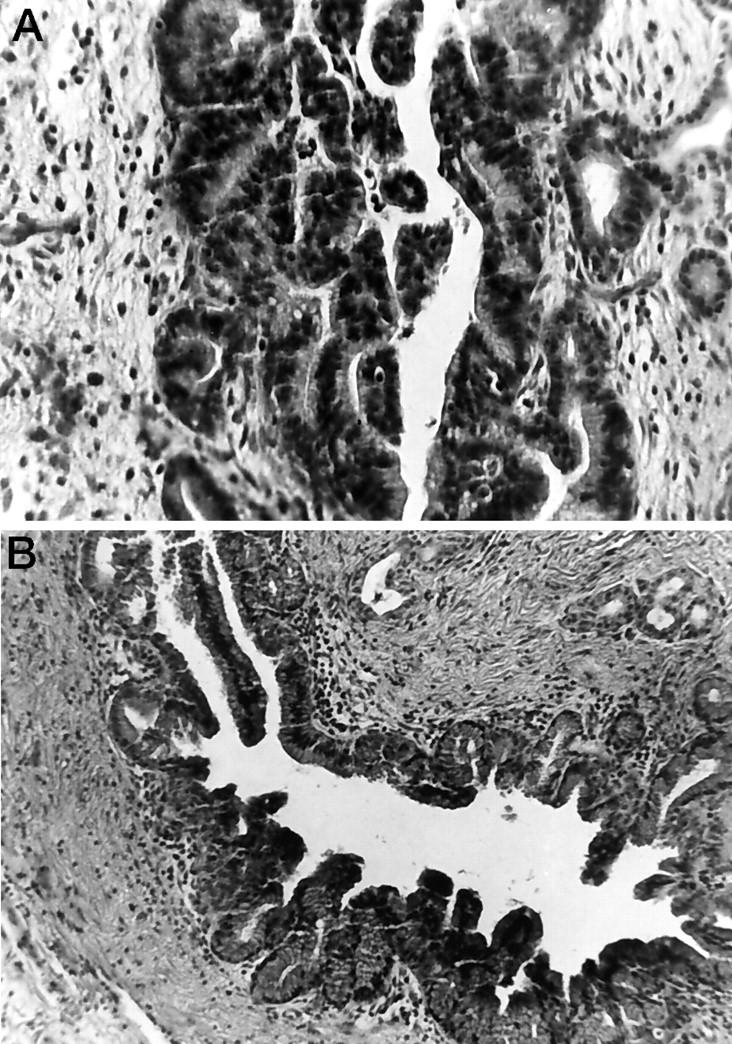
A: The PanIN-3 duct lesion that harbored LOH of the wild-type BRCA2 gene. B: An example of a PanIN-1 duct lesion from patient PX66 that did not have LOH at BRCA2. Hematoxylin and eosin; original magnification, ×40.
In contrast, distinct K-ras mutations were found by sequencing in 7 of the 14 PanINs. All of the K-ras mutations were at codon 12; six had GAT mutations, one was GTT. Results are summarized in Table 1 ▶ . The high signal ratio of mutant to wild-type alleles confirmed the adequacy of the microdissection to exclude non-neoplastic cells. The variety of mutations confirmed the expectations that these lesions would represent independent neoplasms, each with an independent chance to suffer LOH during clonal progression. Thus, the lack of LOH in PX182 (potentially due to a subtle somatic mutation in the other allele) would not preclude the evolution of independent mutations in the PanIN lesions.
Discussion
In this study we provide evidence for the biallelic inactivation of the BRCA2 gene as a late event in the development of adenocarcinoma of the pancreas in patients with germline BRCA2 mutations. Biallelic inactivation of the BRCA2 gene was found in only one high-grade PanIN. This low second hit rate of LOH of BRCA2 in such lesions contrasts with the relative frequency of K-ras mutation and p16 inactivation. 16,24-29 From this data we conclude that BRCA2 inactivation is generally not the first genetic alteration in PanINs from patients with germline BRCA2 mutations.
There are a number of alternative conclusions that could be considered, although we do not find them compelling. First, if the PanIN lesions we studied were not clonal, then LOH would not be expected. Pancreatic cancer is thought to occur through the clonal evolution of precursor lesions. Yet the presence of K-ras mutations in approximately half of the PanINs analyzed supports the clonal nature of these lesions. The prior demonstration of the genetic inactivation of p16 in a subset of PanINs also provided strong genetic evidence that PanINs are the precursor lesions for pancreatic adenocarcinoma and that they are clonal. 28,29 Second, the use of LOH as an indicator of biallelic inactivation does not take into account alternative mechanisms of gene inactivation such as intragenic mutation or promoter methylation. Indeed, some occasional cancers in carriers of BRCA2 mutations lack LOH at BRCA2, and one such cancer is included in the current study. 4 Still, the available evidence points to LOH as the main mechanism of biallelic inactivation of the BRCA2 gene in carcinomas that arise in carriers of BRCA2 mutations. 13 LOH therefore should be present in the vast majority of PanIN if the early biallelic inactivation of BRCA2 were critical to the carcinogenic mechanism. Third, one might consider the low rate of observed LOH in microdissected samples to reflect the contamination of samples with non-neoplastic DNA. This DNA could come from two sources; it could theoretically come from non-neoplastic tissues adjacent to the microdissected foci or cells or DNA other than from the patient. The first potential source of DNA contamination was controlled first by ensuring optimal microdissection using a needle micromanipulator with repeat analysis of all samples and then by confirmation of the expected high rate of K-ras mutations at the expected high allele ratio. The second potential source of DNA would be readily identified on sequencing as the allele ratios would exhibit a predominance of the wild-type allele of BRCA2, yet this was not observed. Finally, an artifact could derive from the use of PEP, which, in the case of low template copy numbers, could introduce a biased amplification of one allele due to stochastic errors (as when the estimated DNA template number is <50 copies). We confirmed that PEP had amplified both alleles of the BRCA2 gene equally among a large panel of samples; hence, an artifact of PEP is not a likely explanation for the single LOH event that we identified in a PanIN sample.
The rarity and apparently late onset of biallelic inactivation of BRCA2 in PanIN contrasts with the timing of APC genetic alterations in colorectal adenomas. Biallelic inactivation of APC is almost certainly the first genetic event leading to the development of adenomas in carriers of a mutant APC gene. 9-12 Another notable difference between the APC gene and the BRCA2 gene is that patients with familial adenomatous polyposis have a greatly increased number of precursor neoplasms; this does not appear to be the case for precursor neoplasms in patients with germline BRCA2 mutations. 30 This suggests that unlike the APC gene in the colon, the BRCA2 gene is not a gatekeeper for neoplasia. An attractive explanation for the rarity of LOH of BRCA2 is that other genetic alterations are required before the biallelic inactivation of BRCA2 can experience favorable selective advantages. In this regard, recent evidence in knockout mouse models suggests that inactivation of the p53 pathway may be such a prerequisite for the survival of embryos that have knockout of BRCA2. 31-34
Two alternate explanations might be entertained. First, epigenetic changes might silence the remaining wild-type gene in some circumstances, Second, BRCA2 may serve a caretaker rather than a strictly suppressor role by participating in chromosome maintenance functions. 35 For example, inherited mutations in caretakers such as the DNA mismatch repair genes produce few precursor lesions and can manifest the tumor phenotype in the absence of allelic deletions. Yet neither possibility is suggested by the accumulated data regarding BRCA2 inactivation in carcinomas, wherein LOH is the prevailing (although not universal) means of inactivation of the remaining wild-type copy of BRCA2.
The relatively late onset of LOH at BRCA2 represents an additional manifestation of the Knudson hypothesis. The fact that biallelic inactivation of a hereditary susceptibility gene is not always the first event in cancer progression may help explain the low penetrance and late age of onset of pancreatic cancer in carriers of BRCA2 mutations 4,5 and the normal number of precursor lesions. Indeed, this paradigm may be a common, yet underappreciated, manifestation of the role of susceptibility genes in cancer predisposition.
Footnotes
Address reprint requests to Scott Kern, M.D., Department of Oncology, 451 Cancer Research Building, The Johns Hopkins Medical Institutions, 1650 Orleans Street, Baltimore, MD 21205-2196. E-mail: sk@jhmi.edu.
Supported by National Institutes of Health SPORE (Specialized Program of Research Excellence) in Gastrointestinal Cancer (CA62924).
References
- 1.Lynch HT, Smyrk T, Kern SE, Hruban RH, Lightdale CJ, Lemon SJ, Lynch JF, Fusaro LR, Fusaro RM, Ghadirian P: Familial pancreatic cancer: a review. Semin Oncol 1996, 23:251-275 [PubMed] [Google Scholar]
- 2.Lynch HT, Fitzsimmons ML, Smyrk TC, Lanspa SJ, Watson P, McClellan J, Lynch JF: Familial pancreatic cancer: clinicopathologic study of 18 nuclear families. Am J Gastroenterol 1990, 85:54-60 [PubMed] [Google Scholar]
- 3.Hruban RH, Petersen GM, Ha PK, Kern SE: Genetics of pancreatic cancer. From genes to families. Surg Oncol Clin North Am 1998, 7:1-23 [PubMed] [Google Scholar]
- 4.Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, Yeo CJ, Jackson CE, Lynch HT, Hruban RH, Kern SE: Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res 1996, 56:5360-5364 [PubMed] [Google Scholar]
- 5.Ozcelik H, Schmocker B, Di Nicola N, Shi XH, Langer B, Moore M, Taylor BR, Narod SA, Darlington G, Andrulis IL, Gallinger S, Redston M: Germline BRCA2 6174delT mutations in Ashkenazi Jewish pancreatic cancer patients. Nat Genet 1997, 16:17-18 [DOI] [PubMed] [Google Scholar]
- 6.Phelan CM, Lancaster JM, Tonin P, Gumbs C, Cochran C, Carter R, Ghadirian P, Perret C, Moslehi R, Dion F, Faucher M-C, Dole K, Karimi S, Foulkes W, Lounis H, Warner E, Goss P, Anderson D, Larsson C, Narod SA, Futreal PA: Mutation analysis of the BRCA2 gene in 49 site-specific breast cancer families. Nat Genet 1996, 13:120-122 [DOI] [PubMed] [Google Scholar]
- 7.Thorlacius S, Olafsdottir G, Tryggvadottir L, Neuhausen S, Jonasson JG, Tavtigian SV, Tulinius H, Ögmundsdottir HM, Eyfjörd JE: A single BRCA2 mutation in male and female breast carcinoma families from Iceland with varied cancer phenotypes. Nat Genet 1996, 13:117-119 [DOI] [PubMed] [Google Scholar]
- 8.Tonin P, Weber B, Offit K, Couch F, Rebbeck TR, Neuhausen S, Godwin AK, Daly M, Wagner-Costalos J, Berman D, Grana G, Fox E, Kane MF, Kolodner RD, Krainer M, Haber DA, Struewing JP, Warner E, Rosen B, Lerman C, Peshkin B, Norton L, Serova O, Foulkes WD, Lynch HT, Lenoir GM, Narod SA, Garber JE: Frequency of recurrent BRCA1 and BRCA2 mutations in Ashkenazi Jewish breast cancer families. Nat Med 1996, 2:1179-1183 [DOI] [PubMed] [Google Scholar]
- 9.Ichii S, Horii A, Nakatsuru S, Furuyama J, Utsunomiya J, Nakamura Y: Inactivation of both APC alleles in an early stage of colon adenomas in a patient with familial adenomatous polyposis (FAP). Hum Mol Genet 1992, 1:387-390 [DOI] [PubMed] [Google Scholar]
- 10.Luongo C, Moser AR, Cledhill S, Dove WR: Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res 1994, 54:5947-5951 [PubMed] [Google Scholar]
- 11.Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW: Inactivation of both APC alleles in human and mouse tumors. Cancer Res 1994, 54:5953-5958 [PubMed] [Google Scholar]
- 12.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AMM, Bos JL: Genetic alterations during colorectal-tumor development. N Engl J Med 1988, 319:525-532 [DOI] [PubMed] [Google Scholar]
- 13.Teng DH, Bogden R, Mitchell J, Baumgard M, Bell R, Berry S, Davis T, Ha PC, Kehrer R, Jammulapati S, Chen Q, Offit K, Skolnick MH, Tavtigian SV, Jhanwar S, Swedlund B, Wong AK, Kamb A: Low incidence of BRCA2 mutations in breast carcinoma and other cancers. Nat Genet 1996, 13:241-244 [DOI] [PubMed] [Google Scholar]
- 14.Hruban RH, Wilentz RE, Goggins M, Offerhaus GJ, Yeo CJ, Kern SE: Pathology of incipient pancreatic cancer. Ann Oncol 1999, 10:9-11 [PubMed] [Google Scholar]
- 15.Cubilla AL, Fitzgerald PJ: Morphological lesions associated with human primary invasive nonendocrine pancreas cancer. Cancer Res 1976, 36:2690-2698 [PubMed] [Google Scholar]
- 16.Klimstra DS, Longnecker DS: K-ras mutations in pancreatic ductal proliferative lesions. Am J Pathol 1994, 145:1547-1550 [PMC free article] [PubMed] [Google Scholar]
- 17.Kozuka S, Sassa R, Taki T, Masamoto K, Nagasawa S, Saga S, Hasegawa K, Takeuchi M: Relation of pancreatic duct hyperplasia to carcinoma. Cancer 1979, 43:1418-1428 [DOI] [PubMed] [Google Scholar]
- 18.Kloppel GBG, Ruckert K, Seifert G: Intraductal proliferation in the pancreas and its relationship to human and experimental carcinogenesis. Virchows Arch A Pathol Anat Histol 1980, 387:221-223 [DOI] [PubMed] [Google Scholar]
- 19.Furukawa T, Chiba R, Kobari M, Matsuno S, Nagura H, Takahashi T: Varying grades of epithelial atypia in the pancreatic ducts of humans: classification based on morphometry and multilvariate analysis and correlated with positive reactions of carcinoembryonic antigen. Arch Pathol Lab Med 1994, 118:227-234 [PubMed] [Google Scholar]
- 20.Longnecker DS: The quest for preneoplastic lesions in the pancreas. Arch Pathol Lab Med 1994, 118:226. [PubMed] [Google Scholar]
- 21.Moskaluk CA, Kern SE: Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. American Journal of Pathology 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N: Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci USA 1992, 89:5847-5851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE: Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res 1998, 58:5329-5332 [PubMed] [Google Scholar]
- 24.Caldas C, Hahn SA, Hruban RH, Redston MS, Yeo CJ, Kern SE: Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res 1994, 54:3568-3573 [PubMed] [Google Scholar]
- 25.DiGiuseppe JA, Hruban RH, Offerhaus GJA, Clement MJ, van den Berg FM, Cameron JL, van Mansfeld ADM: Detection of K-ras mutations in mucinous pancreatic duct hyperplasia from a patient with a family history of pancreatic carcinoma. Am J Pathol 1994, 144:889-895 [PMC free article] [PubMed] [Google Scholar]
- 26.Tabata T, Fujimori T, Maeda S, Yamamoto M, Saitoh Y: The role of Ras mutation in pancreatic cancer, precancerous lesions, and chronic pancreatitis. Int J Pancreatol 1993, 14:237-244 [DOI] [PubMed] [Google Scholar]
- 27.Tada M, Ohashi M, Shiratori Y, Okudaira T, Komatsu Y, Kawabe T, Yoshida H, Machinami R, Kishi K, Omata M: Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology 1996, 110:227-231 [DOI] [PubMed] [Google Scholar]
- 28.Moskaluk CA, Hruban RH, Kern SE: p16 and K-ras mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res 1997, 57:2140-2143 [PubMed] [Google Scholar]
- 29.Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, Yeo CJ, Kern SE, Hruban RH: Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res 1998, 58:4740-4744 [PubMed] [Google Scholar]
- 30.Lakhani SR, Jacquemier J, Sloane JP, Gusterson BA, Anderson TJ, van de Vijver MJ, Farid LM, Venter D, Antoniou A, Storfer-Isser A, Smyth E, Steel CM, Haites N, Scott RJ, Goldgar D, Neuhausen S, Daly PA, Ormiston W, McManus R, Scherneck S, Ponder BA, Ford D, Peto J, Stoppa-Lyonnet D, Easton DF: Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J Natl Cancer Inst 1998, 90:1138-1145 [DOI] [PubMed] [Google Scholar]
- 31.Hakem R, de la Pompa JL, Elia A, Potter J, Mak TW: Partial rescue of Brca1 (5–6) early embryonic lethality by p53 or p21 null mutation. Nat Genet 1997, 16:298-302 [DOI] [PubMed] [Google Scholar]
- 32.Lee H, Trainer AH, Friedman LS, Thistlethwaite FC, Evans MJ, Ponder BA, Venkitaraman AR: Mitotic checkpoint inactivation fosters transformation in cells lacking the breast cancer susceptibility gene, Brca2. Mol Cell 1999, 4:1-10 [DOI] [PubMed] [Google Scholar]
- 33.Ludwig T, Chapman DL, Papaioannou VE, Efstratiadis A: Targeted mutations of breast cancer susceptibility gene homologs in mice: lethal phenotypes of Brca1, Brca2, Brca1/Brca2, Brca1/p53, and Brca2/p53 nullizygous embryos. Genes Dev 1997, 11:1226-1241 [DOI] [PubMed] [Google Scholar]
- 34.Rhei E, Bogomolniy F, Federici MG, Maresco DL, Offit K, Robson ME, Saigo PE, Boyd J: Molecular genetic characterization of BRCA1- and BRCA2-linked hereditary ovarian cancers. Cancer Res 1998, 58:3193-3196 [PubMed] [Google Scholar]
- 35.Mizuta R, LaSalle JM, Cheng HL, Shinohara A, Ogawa H, Copeland N, Jenkins NA, Lalande M, Alt FW: RAB22 and RAB163/mouse BRCA2: proteins that specifically interact with the RAD51 protein. Proc Natl Acad Sci USA 1997, 94:6927-6932 [DOI] [PMC free article] [PubMed] [Google Scholar]