

Abstract
Flavin is one of the most versatile redox cofactors in nature and is used by many enzymes to perform a multitude of chemical reactions. d-Amino acid oxidase (DAAO), a member of the flavoprotein oxidase family, is regarded as a key enzyme for the understanding of the mechanism underlying flavin catalysis. The very high-resolution structures of yeast DAAO complexed with d-alanine, d-trifluoroalanine, and l-lactate (1.20, 1.47, and 1.72 Å) provide strong evidence for hydride transfer as the mechanism of dehydrogenation. This is inconsistent with the alternative carbanion mechanism originally favored for this type of enzymatic reaction. The step of hydride transfer can proceed without involvement of amino acid functional groups. These structures, together with results from site-directed mutagenesis, point to orbital orientation/steering as the major factor in catalysis. A diatomic species, proposed to be a peroxide, is found at the active center and on the Re-side of the flavin. These results are of general relevance for the mechanisms of flavoproteins and lead to the proposal of a common dehydrogenation mechanism for oxidases and dehydrogenases.
D-Amino acid oxidase (DAAO) was one of the first enzymes to be described and the second flavoprotein to be discovered in the mid 1930s (1, 2). It catalyzes the dehydrogenation of d-amino acids to their imino counterparts via the Michaelis complexes M1 and the reduced flavin-product complex M2 (Fig. 1). The reduced flavin is then (re)oxidized by dioxygen to yield FADox and H2O2, whereas the imino acid spontaneously hydrolyzes to the keto acid and NH4+. Although DAAO is present in most organisms and mammalian tissues, its physiological role in vertebrates has been unclear (3). Most recently, however, a specific role of DAAO in the degradation of the neurotransmitter d-serine in brain has been proposed (4), consistent with a role in the regulation of neurotransmission.
Figure 1.

Minimal reaction scheme for DAAO at pH < 8. (a) Binding of the d-amino acid to form the Michaelis complex (M1). (b) Reversible conversion into the reduced enzyme–product complex (M2) involving release of H+. (c) (Re)oxidation by dioxygen, and (d) release of products α-ketoacid and NH4+.
The dehydrogenation, catalyzed by the class of flavoprotein oxidases and dehydrogenases, as exemplified by DAAO, is a fundamental biochemical reaction. Despite this, its molecular mechanism is still a matter of dispute and has evoked several contrasting proposals over the years. In 1971, Walsh et al. (5) discovered that pig kidney DAAO (pkDAAO) catalyzes the elimination of halide from β-halogenated amino acids. This led to the seemingly reasonable conclusion, found in most biochemistry textbooks, that catalysis involves abstraction of the amino acid αH as H+ via the so-called “carbanion mechanism,” a process that requires an active site base. This concept was challenged in 1975, based on work with pkDAAO reconstituted with the artificial cofactor 5-deazaFAD by Hersh and Schuman-Jorns (6), who favored a hydride mechanism proceeding via transfer of the substrate αC—H to the flavin N (5). These mechanisms, the carbanion and the hydride transfer, represent the two extremes under consideration for such a chemical oxidation reaction.
Recently the three-dimensional structure of pkDAAO complexed with benzoate, iminotryptophane, and o-aminobenzoate at 2.5- to 3.0-Å resolution was reported by two independent groups (7–10). The mechanistic interpretations of the structural data were quite contrasting. On the basis of molecular modeling of d-leucine into the active site of pkDAAO, Miura et al. (10) proposed an electron–proton–electron mechanism in which “… two electrons flow from the amino lone-pair in concert with the α-proton abstraction by N(5) of flavin.” This formulation could explain β-halide elimination. On the other hand, Mattevi's group favored a classical hydride mechanism based on the mode of interaction of substrate (d-alanine) modeled into the active site of pkDAAO (7). This latter hypothesis is supported by linear free energy correlations, which indicate that no significant charge develops in the transition state of the reaction catalyzed by Trigonopsis variabilis DAAO (11). Kinetic isotope effects are consistent with a concerted cleavage of the substrate C—H and N—H bonds in the catalytic reaction and argue against the occurrence of intermediates (11). Here we report the very high-resolution three-dimensional structure of DAAO from the yeast Rhodotorula gracilis (RgDAAO) complexed with d-alanine, d-trifluoroalanine (d-F3-Ala), and l-lactate. The relative orientation of the flavin cofactor and of the substrate/ligands is unequivocally in favor of a hydride mechanism as proposed by Mattevi et al. (7).
Materials and Methods
Protein Preparation, Spectral Characterization, Crystallization, Data Collection, and Reduction.
Recombinant RgDAAO was obtained as described previously (12). Kds for ligands were measured spectrophotometrically with 1-ml samples of ≈10 μM enzyme in 50 mM Na pyrophosphate buffer, pH 7.5 (+ 1%, vol/vol glycerol), at 15°C. Spectral changes were followed at 345 (l-lactate) and 485 nm (d-F3-Ala). Crystals were grown with the hanging drop vapor diffusion technique by mixing 4 μl of concentrated protein in 20 mM Hepes buffer, pH 7.5, with an equal volume of reservoir solution (100 mM Hepes, pH 7.5/15% polyethylene glycol 10,000/200 mM ammonium sulfate containing 1 mM l-lactate). Crystals grew within 5 days to a final size of approximately 350 × 350 × 150 μm at 18°C. For the d-Ala complex, a crystal was soaked with 20 mM d-alanine in the presence of 200 mM pyruvate. During this procedure, the crystal became colorless and remained so during data collection. It diffracted to 1.2 Å at the ×11 beam line at Deutsches Elektronen Synchrotron (DESY), Hamburg. The d-F3-Ala crystal was prepared by soaking with a mixture of 10 mM dl-F3-Ala. Data from this crystal were measured to 1.72 Å resolution at a rotating anode x-ray source (Schneider, Offenburg, Germany) with a MAR345 image plate system. Data from RgDAAO complexed with l-lactate were collected to 1.47 Å at the BW7B beam line at DESY. All crystals were flash frozen in the presence of 20% glycerol as a cryoprotectant. Data reduction and space group determination were done by using xds (13).
Molecular Replacement and Refinement.
Rotation and translation functions were calculated with cns (14) by using pkDAAO (Protein Data Bank code 1AA8) (10) as a search model, and an unambiguous solution was found. The resulting electron density maps were of sufficient quality to model the correct protein sequence and the FAD cofactor. The model was refined with CNS by using standard protocols for simulated annealing, minimization, and B factor refinement. At an R factor of 20.4%, refinement was continued with shelxl (15). The model obtained from refinement with the 1.47 Å data set was used as a starting point for the refinement of the 1.20 Å and 1.72 Å data sets (Table 1). H-atom positions were derived from the positions of C, N, O, and S atoms by shelxl and refined in their “riding” geometry against the 1.20- and 1.47-Å data sets. Geometry restraints in refinement were those by Engh and Huber (17) for the protein or were obtained from coordinates deposited in the Cambridge Structure Database for the ligands. Water molecules were assigned, and their occupancy was refined with shelxl if they appeared in difference electron density maps and were also visible in 2Fo − Fc maps. For the structures at 1.20 Å and 1.46 Å, no restraints for FAD were implemented in shelxl. For the identification of the ligand(s) in the 1.20-Å structure, the following procedure was used: (i) pyruvate with restraints on its planar configuration was first modeled into the electron density. The resulting fit of the model and the electron density were unsatisfactory. (ii) d-Ala without tetrahedral restraints gave a good fit to the electron density. (iii) A mixture of d-Ala and pyruvate as alternate ligands with a total occupancy of 100% did not improve the fit substantially but yielded estimates for the occupancy of d-Ala and pyruvate of 80 and 20%, respectively. The bond angles found around the ligand αC center are: 114.3° (Cα,N,CH3), 112.2° (Cα,N,COOH), 119.3° (Cα,COOH,CH3). A full description of the structures will be presented elsewhere (unpublished work).
Table 1.
Crystallographic data and refinement statistics
| Diffraction data | Ligand
|
||
|---|---|---|---|
| d-Alanine | d-F3-Ala | l-Lactate | |
| Redox state of flavin | Reduced | Oxidized | Oxidized |
| Resolution limits | 100–1.20 | 100–1.72 | 100–1.47 |
| Wavelength, Å | 0.9114 | 1.5418 | 0.8345 |
| Observed reflections | 1,000,779 | 230,642 | 515,122 |
| Unique reflections | 155,642 | 52,652 | 83,893 |
| Completeness, % | 99.9 (99.9) | 98.9 (96.7) | 99.1 (91.2) |
| Rmrg-F (35) | 8.4 (42.4) | 11.0 (37.2) | 5.3 (28.8) |
| I/σ | 15.7 (2.7) | 8.9 (2.3) | 20.1 (3.2) |
| Final refinement statistic* | |||
| Final R factor/R-free*, % | 11.8/15.0 | 16.2/20.3 | 11.2/16.1 |
| Anisotropic B factor refinement | All nonhydrogens atoms | Sulfur and phosphate | All nonhydrogen atoms |
| Hydrogens | included | not included | included |
| No. of disordered atoms† | 38 | 14 | 24 |
| Solvent molecules, full occupancy | 613 | 580 | 648 |
| Solvent molecules, half occupancy | 68 | 137 | 141 |
| Model rms deviations from ideality‡ | |||
| Bond lengths, Å | 0.008 | 0.011 | 0.008 |
| Bond angles, ° | 1.57 | 2.48 | 1.73 |
Parentheses denote the highest shell. Rmrg-F = Σ | Ihkl − 〈Ihkl〉 |/〈Ihkl〉, where Ihkl is the intensity of an observation of reflection hkl, and 〈Ihkl〉 is the average intensity for reflection hkl.
The same test set of reflections was used for calculating Rfree (16) in all refinements.
For disordered residues, only the major component atoms were considered.
Calculations performed with shelxpro (17).
Results
Structure of RgDAAO in the Presence of Active-Site Ligands.
d-Alanine was selected as ligand, because it is one of the best substrates and binds reasonably tightly (Kd ≈ 3.0 ± 0.8 mM at pH 8.5 and 25°C) (18). Its reaction with (yellow) oxidized flavin was expected to yield the (colorless) complex of reduced flavin with the product iminopyruvate and corresponding information. F3-Ala is, from a structural point of view, closely similar to alanine; in contrast to d-Ala, however, d-F3-Ala does not reduce the enzyme flavin in static experiments (19). This is because of the inductive effect of the CF3 group, which causes a shift of the equilibrium of step (b) in Fig. 1 from M2 completely toward M1. Binding of d-F3-Ala to RgDAAO (Kd = 6.2 ± 0.6 mM) is similar to that of d-alanine, and thus formation of the Michaelis complex M1 was anticipated. The d-F3-Ala should yield information on the relative orientation of substrate and oxidized flavin. From the structure of the l-lactate complex (Kd = 16.2 ± 1.4 mM), information complementary to that arising from the d-enantiomeric ligands was expected.
The structure of RgDAAO colorless crystals assumed to contain reduced flavin was solved and refined at 1.20 Å (Fig. 2), at which resolution differences in the number of electrons between carbon, nitrogen, and oxygen can be visualized (Figs. 3, 4, and 5). Electron densities for hydrogens are observed at defined positions such as the flavin N(3), C(6), and C(9) atoms (sp2 hybrids), as well as C(1′) and C(7) (sp3 hybrids) (Fig. 5). Two crucial issues in the context of this structure are the nature of the ligand and the oxidation state of the flavin. The latter would be defined by the presence of N(5)—H in the electron density. Visualization of electron density differences corresponding to less than one electron, however, is at the limit of the experimental error. For these reasons and because of H-inversion at the N(5) pyramidal center, which reduces the occupancy on each of the two (Si and Re) sides of the reduced flavin plane to 0.5, this is not possible (Fig. 5). On the other hand, the following findings support the presence of predominantly reduced flavin: the crystals remain colorless until completion of data collection, and the flavin is thus, at least to a major extent, in its reduced form. The lengths of specific bonds inside the isoalloxazine are different from those found for the complexes with l-lactate and d-F3-Ala, which contain oxidized flavin. The largest difference is found for the length of the N(5)—C(4a) bond, which also exhibits the largest changes on reduction in small-molecule x-ray structures of flavin models (20). In the d-Ala complex, the N(5)—C(4a) length is 1.477 (±0.014) Å. This is consistent with the length of the N(5)—C(4a) single bond reported for the reduced flavin model (1.418 Å) and compares to 1.300 Å for the N(5)⩵C(4a) double bond in the oxidized model (20). The presence of d-Ala and oxidized flavin at the active center is also mutually exclusive.
Figure 2.
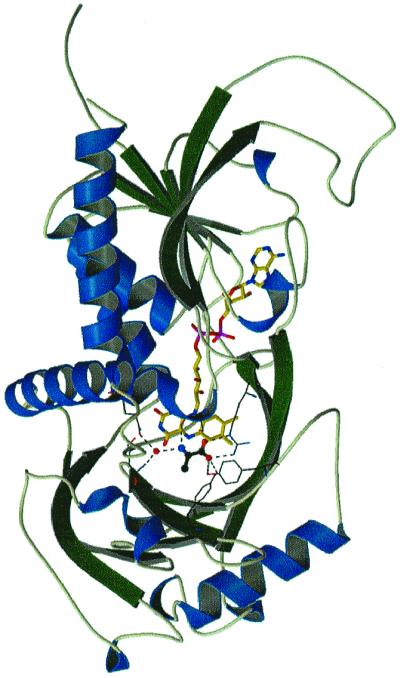
General representation of RgDAAO structure. The substrate d-alanine is located above the reduced flavin Re-side. (– – –) lines denote hydrogen bonds involved in substrate fixation.
Figure 3.

Active site of RgDAAO at 1.2-Å resolution. Stereo view of the 2Fobs − Fcalc map (orange, 3σ) and the omit map (magenta, 3σ) showing clear electron density assigned to a peroxide species. The data were obtained from RgDAAO crystals soaked with 20 mM d-alanine and 200 mM pyruvate.
Figure 4.

Reciprocal orientation of ligands and flavin plane. For clarity, the dioxygen species has been omitted. Dashed lines represent H-bonds. (A) d-Ala is viewed along its N-αC axis; the electron density is shown at 2σ. (B) d-F3-Ala (Upper) and l-lactate (Lower). The green trace represents the ideal line connecting the flavin N(5) and the ligand αC centers (distance ≈ 3.2 Å). Note that the αC—H function (grey) of l-lactate points away from the flavin. With l-lactate, the position of the α—H results from H-inclusion in the refinement. In the case of d-F3-Ala (A), the number of observations (1.72 Å) does not allow positioning. The strong H-bond interactions with the α-NH2/OH, together with the electrostatic interaction of the substrate carboxylate group with Arg-285, Tyr-238, and Tyr-223, provide the rationale for substrate d-specificity in that they prevent binding of the l-amino acid in a productive manner.
Figure 5.

Evidence for hydrogens and a peroxide species. The isoalloxazine ring of the flavin is seen from the Re-side together with its 2Fobs − Fcalc map (orange, 3σ). Notice the orientation of the postulated peroxide molecule (in red) and its electron density (3σ). Additional omit density (3σ) is shown in green for the H-atoms. Clear density is visible for N(3), C(6), C(9), and C(1′) hydrogens, whereas the hydrogens at N(5), C(7)—CH3 and C(8)—CH3 are diffuse or not observable.
For the ligand, alanine or (imino)pyruvate is the species to be considered. As shown in Fig. 4A, both the αC and αN atoms have significant tetrahedral character, although the angles between the bonds (see Materials and Methods) do not correspond to the values of an ideal tetrahedron. In (imino)pyruvate, both centers are planar (sp2 configuration), whereas for d-Ala, a tetrahedral (sp3) configuration would be expected as is found, e.g., for l-lactate (Fig. 4B). The length of the αC—N bond is 1.39 ± 0.01 Å. Comparison of this value to bond lengths of 1.47 Å in alanine (21) and 1.21 Å for the carbonyl double bond in pyruvate (22) suggests an occupancy of d-Ala and (imino)pyruvate in a ratio ≥2:1. Despite the fact that d-Ala is not the sole ligand at the active center, we conclude that the mode of binding as shown in Fig. 4A is unequivocal with respect to the position of the αC—H bond, which is pointing toward the flavin plane.
The mode of binding of the ligands (Figs. 3 and 4) follows the pattern found with flavocytochrome b2, glycollate oxidase, and pkDAAO (7): (i) the carboxylate is anchored via an electrostatic interaction to the Arg-285 guanidinium group (distances 2.76 and 2.77 Å); (ii) one of the carboxylate oxygens forms H-bonds with Tyr-223—OH (2.68 Å) and Tyr-238—OH (3.36 Å); (iii) the substrate α-NH3+ is H-bonded symmetrically with Ser-335⩵O and Wat-72 (both at 2.82 Å), the latter groups serving as acceptors. Wat-72 is fixed in place by Asn-54 Nδ and Gln-339 Oɛ. None of these latter residues is in contact with groups capable of acid-base catalysis. The ligand side chain is oriented toward a hydrophobic binding pocket. This arrangement contrasts with that of pkDAAO, where only Tyr-228 (corresponding to Tyr-223 in RgDAAO) forms one H-bond to the substrate —COO− (7, 9). In RgDAAO, both Tyr-223 and Tyr-238 thus serve in substrate binding/fixation and do not play a functional role in chemical catalysis. In fact, mutation of each of these two Tyr to Phe does not abolish dehydrogenase activity but impairs ligand binding (19).
In all three structures (Figs. 3 and 4), the difference in the positions occupied by the ligand α-function (α-NH3+ or α-OH) and the carboxylate oxygens is less than 0.2 Å. The position of the three observable αC substituents of d-Ala and d-F3-Ala define the direction of the αC—H bond, which thus points toward the (oxidized) flavin N(5) atom, i.e., its lowest unoccupied molecular orbital (LUMO) (Fig. 4). The same mode of fixation of the carboxylate and α-OH functions is retained with l-lactate, which leads to the opposite orientation of the αC—H, i.e., to point away from the flavin.
No particular differences are observed between the structures of the enzyme in the oxidized (d-F3-Ala and l-lactate complexes) and in the reduced state (d-Ala complex). Specifically, the oxidized and reduced isoalloxazines and corresponding functional groups at the active site occupy identical positions (Fig. 4), and the isoalloxazine moiety is essentially planar in all complexes. From this, we conclude that structural information obtained from the oxidized and reduced enzyme forms can be used interchangeably.
Identification of a Diatomic Species, Possibly Derived from Dioxygen, at the Active Center.
The high-resolution electron density of the complex with d-alanine permits the observation of a diatomic species with partial occupancy at the active center. A dioxygen species can be modeled into the electron density and refined to ≈20% occupancy (Figs. 3 and 5). This corresponds to 1.6 electrons (20% of 8 electrons at each atomic position) and is therefore still defined within the limits of the experimental error. The electron density does not allow a direct identification of the molecular species. O2, or a peroxide structural element, all of which might be involved in the reoxidation reaction, are possible candidates. The distance between the atom positions after unrestrained refinement is 1.56 (±0.04) Å, suggesting a peroxide moiety (theoretical O–O bond distance ≈1.5 Å, compared with 1.18 Å for O2). One atom of the species is placed on the flavin plane between N(5) and (4a) (Fig. 5) [distances: 1.61 Å to N(5), 1.78 Å to C(4a), 2.03 Å to C(5a), and 2.39 Å to C(10a)]. The second atom is at similar distance from the flavin plane [1.80 Å to C(5a) and 1.78 Å C(9a)]. This argues for a covalent bond with the flavin and against a complex with, e.g., H2O2, because in the latter case a distance >2.5 Å should be expected. For the same reason, the presence of two mutually exclusive molecules of H2O, each with fractional occupancy of 20%, is unlikely: the water molecules would be placed above the flavin plane, and their total occupancy would be 40%. Their presence is sterically incompatible with that of the ligand d-Ala/iminopyruvate [Ala-Cα to N(5) ≈3.2 Å, Ala-CH3 to C(4a)≈3.4 Å], it thus would lower the total occupancy of the latter to a value which is incompatible with the crystallographic data.
Discussion
Mechanism of Substrate Dehydrogenation by DAAO.
Two observations are crucial for mechanistic interpretations: first, the orientations of both d-alanine and d-F3-Ala, with the αC—H function pointing toward the flavin N(5), are exactly what is required for efficient hydride transfer (Figs. 3 and 4). Second, no functional group is present at the active center that might act as an acid-base catalyst in substrate αC—H bond cleavage. Under these circumstances, a carbanion mechanism can be excluded, because its classical formulation cannot be envisaged in the absence of a base functioning in H+ abstraction. This situation is reminiscent of that encountered in NAD(P)H dehydrogenases, where hydride transfer occurs within the complex between flavin and pyridine nucleotide, and where involvement of functional groups in the hydride transfer step is also not evident.
From these data, a mechanistic picture emerges that is striking in its simplicity (Fig. 6). At optimal high pH, the Michaelis complex M1 contains amino acid with neutral α-NH2 group. The orbital of the substrate αC—H is aligned with the lowest unoccupied molecular orbital (LUMO) of the flavin N(5), with which it can overlap, and the α-NH2 group is placed at ≈3.2 Å above the flavin positions C(4a)—C(4) (Figs. 3–5). The ensuing hydride transfer is coupled with the transformation of the tetrahedral substrate into a planar product and leads directly to complex M2 (Fig. 1). During this process, the incipient negative charge is delocalized on the pyrimidine ring of the flavin and is stabilized by the positive charge generated on the product αC⩵NH2+ group (Fig. 6). The interplay of these two charges is assumed to be an important factor in governing the thermodynamics of the system. The complex of reduced anionic (23) flavin with (zwitterionic) d-alanine (α-NH3+) can be regarded as a mimic of this model.
Figure 6.

Schematic representation of substrate interactions at the active site and mechanisms of dehydrogenation via hydride transfer. The absolute configuration of the substrate α-substituents is undefined. d-Ala is bound in its zwitterionic form (αNH3+). Note that the negative charge on the reduced flavin is balanced by the positive one on the product (α⩵NH2+), and that concomitantly with hydride transfer to the flavin, one H+ is transferred to the solvent. The top and bottom species correspond to the Michaelis complexes M1 and M2 shown in Fig. 1.
Agreement Between Structural and Kinetic Data.
The mechanism we derive from these data (Fig. 6) is in agreement with the pH dependence of the rate of substrate dehydrogenation. Thus, from the pH dependence of kcat/Km with phenylglycine, an apparent pKa ≈8.1 was estimated, indicating that one of the groups involved is deprotonated at pH > pK for efficient dehydrogenation (11). In combination with the present structural data that exclude the presence of amino acid functional groups with such properties at the active center, we attribute this pK to the substrate α-NH2 group.
The proposed mechanism also agrees with the observation of large kinetic deuterium isotope effects at pH ≤ 7 on the rate of flavin reduction measured in stopped-flow experiments with d-Ala as substrate. These are 3.1 ± 1.1 for the solvent [d-[2-1H]Ala in 1H2O/2H2O] and 9.1 ± 1.5 for the primary one (d-[2-1H]Ala/d-[2-2H]Ala). The “double-isotope effect” [d-[2-1H]Ala in 1H2O vs. d-[2-2H]Ala in 2H2O] is 26 ± 8 at pH < 7. At pH > 8, the primary isotope effect with d-Ala is decreased to ≈2.35 and the solvent one to ≈1.25 (C. M. Harris, L.P. and S.G., unpublished observations). Similar isotope effects have been reported for the T. variabilis DAAO and the substrate phenylglycine (11). This indicates that at pH ≤ 7, where the amino acid is in the α-NH3+ (zwitterionic) form (Michaelis complex M1, Figs. 1 and 6), the large isotope effect found results from a multiplication of two single-isotope effects in a concerted or synchronous process (11); i.e., hydride transfer is coupled with deprotonation of the α-NH3+ in a single transition state. Indeed, Fitzpatrick and Massey (23) have shown that one H+ is released concomitantly with flavin reduction (Fig. 1b) during the anaerobic reaction of pkDAAO with d-Ala. We propose that this H+ originates from the amino acid α-NH3+. [A second H+ is released in the subsequent step, the iminoacid dissociation (23).] At pH > pK of the substrate α-NH3+ in M1, the (smaller) isotope effect results from fission of solely the αC—H bond.
These results raise the question about the mode of transfer of H+ from the α-NH3+ to solvent. As shown in Fig. 4A, Ser-335⩵O and Wat-72 are placed to accept H-bonds from the α-N and are slightly displaced toward the flavin side. The geometry of Ser-335⩵O and Wat-72 is such that the H-bond strengths would increase during the transition from substrate (sp3) to the planar transition state and product (sp2). This geometry also suggests that the third proton of the α-NH3+, probably the one being removed, is placed in a trans position with respect to the αC—H. Although the N—H to be released is oriented toward bulk solvent, the α-NH3+ is separated from it by Ser-335—OH, which is at a distance of 4.75 Å and is in contact with Wat-457, Wat-285, and Wat-88, these in turn being in contact with solvent. By simple rotation around the αC—βC bond, Ser-335—OH can be brought within H-bond distance to α-NH3+ and in the position required for interaction with the anticoplanar H of the latter. This geometry would be perfect for a concerted trans-elimination reaction optimal for hydride transfer, and Ser-335—OH would thus serve in the translocation of H+ to bulk solvent. This is in agreement with preliminary results obtained with the Ser-335→Gly mutant of RgDAAO. At pH 8 and with d-Ala, the Ser-335→Gly mutation does not alter significantly the catalytic parameters compared with wild-type RgDAAO. At pH 6.0, however, kcat and the rate of flavin reduction are ≈1/10 those of wild type (unpublished results). Similarly, in the recently solved structure of snake venom l-amino acid oxidase, the active-center His-223 is found to occupy two alternative positions (24). One corresponds to that required for an interaction with the anticoplanar H of the l-substrate α-NH3+. In the second, it “swings away” by rotation around the αC—βC bond to a position similar to that of Ser-335—OH in RgDAAO, shown in Fig. 4A.
General Mechanisms of Substrate Dehydrogenation and Dioxygen Activation.
d-lactate is a substrate of both pkDAAO (25) and RgDAAO, albeit a very slow one (flavin reduction: t1/2 ≈ 250 min with 100 mM d-lactate at 25°C; unpublished observations). A comparison of flavin enzymes dehydrogenating α-OH and α-amino acids is thus instructive and leads to the formulation of a common mechanism. These two subclasses of enzymes have long been assumed to be related, and the finding of major differences in the nature and presence of functional groups at the active centers has been puzzling (7, 9). In flavocytochrome b2 and glycollate oxidase (26, 27), a lysine-NH3+ close to the flavin pyrimidine N(1) stabilizes the charge developing on the flavin on reduction. In RgDAAO, such a group is absent; however, we propose that the same stabilization is exerted by the product α⩵NH2+. Arg-285 appears to be involved in the stabilization of negatively charged flavin in the absence of ligands (28). It should be noted that with DAAOs, an (additional) stabilization of negatively charged flavin is observed also in the presence of carboxylate ligands that neutralize Arg-285 (3).
In flavocytochrome b2 and glycollate oxidase, a His (in charge relay contact with an Asp) was originally proposed to function in the abstraction of the substrate αC—H as H+ (26, 27). However, this His could serve in the abstraction of the weakly acidic αC—OH as H+ (pKa for lactate αC—OH ≈15), thus initiating the expulsion of hydride (29). This would correspond to the H+ relay system (base pair) functioning in H+ abstraction from C—OH in the dehydrogenation catalyzed by pyridine nucleotide dependent lactate dehydrogenases (30). Such a system is absent in RgDAAO. At high pH (≥8), the substrate amino acid binds in the “α-NH2 form” that is suitable for immediate hydride transfer. At pH ≤ 7 (α-NH3+ form), the H+ liberated during hydride transfer is proposed to be relayed to bulk solvent via an H-bridge to Ser-335—OH. In pkDAAO, an analogous function could be envisaged for Tyr-224 and in l-amino acid oxidase for His-223 (24).
The pKa of α-NH3+ is ≈6 units lower compared with that of a corresponding α-OH in a carboxylic acid. Consistent with this difference, the rate of d-Ala dehydrogenation by RgDAAO at pH 8.5 is seven orders of magnitude higher than that for d-lactate. These considerations suggest that dehydrogenation of α-amino and of α-hydroxyacids by flavoproteins proceeds by the same hydride transfer mechanism (see Fig. 6). It should be stressed, however, that an evident dilemma exists in that a hydride transfer mechanism cannot rationalize the elimination reactions observed with both classes of enzymes. Although alternate mechanisms cannot be discussed here in detail, we hypothesize that elimination proceeds via secondary reactions as suggested earlier (31). In view of the absence of base functional groups, it is most unlikely that this occurs at the level of oxidized flavin with DAAO. An elimination starting at the level of reduced enzyme flavin/iminoacid complex is a more probable alternative (31).
With flavin monooxygenases, dioxygen activation has been shown to proceed via C(4a) covalent hydroperoxides (32), which are remarkably stable. With oxidases, the identification of such species has been elusive, the assumed reason being their kinetic instability. In our structure, the atomic distances would be compatible with a dioxygen species bound to either the flavin positions N(5) or C(4). Chemical arguments and the analogy to monooxygenases would rather point toward a link to C(4a).
In conclusion, the present results add strong arguments in favor of the hydride transfer mechanism in its classical formulation (33) and fully support the proposal of Mattevi et al. (7). This mechanism appears to be uncommon in that the chemical transformations can proceed efficiently without the involvement of amino acid functional groups. Most groups present at the active site are involved in substrate recognition, binding, and fixation, i.e., they direct the trajectory of the interacting orbitals. This ability is a primary constituent of the catalytic power of DAAO. Hydride transfer is accompanied by bond reorganization and transformation of the tetrahedral substrate αC-center into a planar product. The present mechanism would represent a mode of catalysis in which orbital steering/interactions are the predominant factors for the chemical step(s). It would be in line with the concepts, discussed recently by Koshland's group (34), that orbital orientation is a major quantitative factor in enzyme catalysis.
Acknowledgments
We are indebted to one referee for very helpful suggestions. This research was supported by a grant from Ministero dell'Università e della Ricerca Scientifica e Tecnologica to M.S.P. (PRIN 9705182517-018).
Abbreviations
- DAAO
d-amino acid oxidase
- pkDAAO
pig kidney DAAO
- d-F3-Ala
d-trifluoroalanine
- RgDAAO
Rhodotorula gracilis DAAO
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates reported in this paper have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1c0p, 1c0l, and 1c0k).
References
- 1.Krebs H A. Biochem J. 1935;29:1620–1625. doi: 10.1042/bj0291620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warburg O, Christian W. W Biochem Z. 1938;298:150–155. [Google Scholar]
- 3.Curti B, Ronchi S, Pilone Simonetta M. In: Chemistry and Biochemistry of Flavoenzymes. Müller F, editor. III. Boca Raton, FL: CRC; 1992. pp. 69–94. [Google Scholar]
- 4.Wolosker H, Blackshaw S, Snyder S H. Proc Natl Acad Sci USA. 1999;96:13409–13414. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh C T, Schonbrunn A, Abeles R. J Biol Chem. 1971;248:6855–6866. [PubMed] [Google Scholar]
- 6.Hersh L B, Schuman-Jorns M. J Biol Chem. 1975;250:8728–8734. [PubMed] [Google Scholar]
- 7.Mattevi A, Vanoni M A, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B. Proc Natl Acad Sci USA. 1996;93:7496–7501. doi: 10.1073/pnas.93.15.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Todone F, Vanoni M A, Mozzarelli A, Bolognesi M, Coda A, Curti B, Mattevi A. Biochemistry. 1997;36:5853–5860. doi: 10.1021/bi9630570. [DOI] [PubMed] [Google Scholar]
- 9.Mizutani H, Miyahara I, Hirotsu K, Nishina Y, Shiga K, Setoyama C, Miura R. J Biochem. 1996;120:14–17. doi: 10.1093/oxfordjournals.jbchem.a021376. [DOI] [PubMed] [Google Scholar]
- 10.Miura R, Setoyama C, Nishina Y, Shiga K, Miyutani H, Miyuhara I, Hirotsu K. J Biochem. 1997;122:825–833. doi: 10.1093/oxfordjournals.jbchem.a021829. [DOI] [PubMed] [Google Scholar]
- 11.Pollegioni L, Blodig W, Ghisla S. J Biol Chem. 1997;272:4924–4934. doi: 10.1074/jbc.272.8.4924. [DOI] [PubMed] [Google Scholar]
- 12.Molla G, Vegezzi C, Pilone M S, Pollegioni L. Protein Expression Purif. 1998;14:289–294. doi: 10.1006/prep.1998.0956. [DOI] [PubMed] [Google Scholar]
- 13.Kabsch W J. J Appl Crystallogr A. 1988;50:157–163. [Google Scholar]
- 14.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J-S, Kuszewski J, Nilges M, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 15.Sheldrick G M, Schneider T R. In: Methods in Enzymology. Carter C W Jr, Sweet R M, editors. Vol. 277. New York: Academic; 1997. pp. 319–343. [PubMed] [Google Scholar]
- 16.Brünger A T. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 17.Engh R A, Huber R. Acta Crystallogr. 1991;A 47:392–400. [Google Scholar]
- 18.Pollegioni L, Langkau B, Tischer W, Ghisla S, Pilone M S. J Biol Chem. 1993;268:13850–13857. [PubMed] [Google Scholar]
- 19.Harris C M, Molla G, Pilone M S, Pollegioni L. J Biol Chem. 1999;274:36233–36240. doi: 10.1074/jbc.274.51.36233. [DOI] [PubMed] [Google Scholar]
- 20.Porter D J T, Voet D. Acta Crystallogr B. 1978;34:598–606. [Google Scholar]
- 21.Fletterick R J, Tsai C, Hughes R H. J Phys Chem. 1971;75:918–922. [PubMed] [Google Scholar]
- 22.Kratochvil B, Ondracek J, Krechl J, Hasek J. Acta Crystallogr C. 1987;43:2182–2183. [Google Scholar]
- 23.Fitzpatrick P F, Massey V. J Biol Chem. 1982;257:9958–9962. [PubMed] [Google Scholar]
- 24.Pawelek P, Cheah J, Coulombe R, Macheroux P, Ghisla S, Vrielink A. EMBO J. 2000;19:4204–4215. doi: 10.1093/emboj/19.16.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yagi K, Ozawa T, Naoi M, Kotaki A. In: Flavins and Flavoproteins. Yagi K, editor. Tokyo: Univ. of Tokyo Press; 1968. pp. 237–251. [Google Scholar]
- 26.Lederer F. In: Chemistry and Biochemistry of Flavoenzymes. Müller F, editor. II. Boca Raton, FL: CRC; 1991. pp. 153–242. [Google Scholar]
- 27.Xia Z-X, Shamala N, Bethge P H, Lim L W, Bellamy H D, Xuong N H, Lederer F, Mathews F S. Proc Natl Acad Sci USA. 1987;84:2629–2633. doi: 10.1073/pnas.84.9.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molla G, Porrini D, Job V, Motteran L, Vegezzi C, Campaner S, Pilone M S, Pollegioni L. J Biol Chem. 2000;275:24715–24721. doi: 10.1074/jbc.M908193199. [DOI] [PubMed] [Google Scholar]
- 29.Steinberg K, Clausen T, Lindqvist Y, Macheroux P. Eur J Biochem. 1995;228:408–416. [PubMed] [Google Scholar]
- 30.Abad-Zapatero C, Griffith J P, Sussman J L, Rossmann M G. J Mol Biol. 1987;198:445–467. doi: 10.1016/0022-2836(87)90293-2. [DOI] [PubMed] [Google Scholar]
- 31.Massey V, Ghisla S, Ballou D P, Walsh C T, Cheung Y T, Abeles R H. In: Flavins and Flavoproteins. Singer T S, editor. Amsterdam: Elsevier; 1975. pp. 199–212. [Google Scholar]
- 32.Ghisla S, Massey V. Eur J Biochem. 1989;1811:1–17. doi: 10.1111/j.1432-1033.1989.tb14688.x. [DOI] [PubMed] [Google Scholar]
- 33.Kohen A, Klinman J P. Chem Biol. 1999;6:191–198. doi: 10.1016/s1074-5521(99)80058-1. [DOI] [PubMed] [Google Scholar]
- 34.Mesecar A D, Stoddard B L, Koshland D E., Jr Science. 1997;277:202–206. doi: 10.1126/science.277.5323.202. [DOI] [PubMed] [Google Scholar]
- 35.Diederichs K, Karplus A. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]