

Abstract
Deposition of aggregated protein into neurofilament-rich cytoplasmic inclusion bodies is a common cytopathological feature of neurodegenerative disease. How—or indeed whether—protein aggregation and inclusion body formation cause neurotoxicity are presently unknown. Here, we show that the capacity of superoxide dismutase (SOD) to aggregate into biochemically distinct, high molecular weight, insoluble protein complexes (IPCs) is a gain of function associated with mutations linked to autosomal dominant familial amyotrophic lateral sclerosis. SOD IPCs are detectable in spinal cord extracts from transgenic mice expressing mutant SOD several months before inclusion bodies and motor neuron pathology are apparent. Sequestration of mutant SOD into cytoplasmic inclusion bodies resembling aggresomes requires retrograde transport on microtubules. These data indicate that aggregation and inclusion body formation are mechanistically and temporally distinct processes.
Familial amyotrophic lateral sclerosis (FALS), a dominantly inherited form of ALS, is a progressive paralytic disorder resulting from the degeneration of motor neurons in the cortex, brainstem, and spinal cord (1, 2). Between 10% and 20% of FALS cases are because of missense mutations in the SOD1 gene, which encodes the cytoplasmic metalloenzyme Cu, Zn-superoxide dismutase (SOD) (3). Mice expressing human FALS-linked SOD1 transgenes develop an age-dependent ALS-like disorder characterized by profound degeneration of spinal motor neurons and by the presence in surviving motor neurons of neurofilament-rich cytoplasmic inclusions resembling pathological inclusion bodies in spinal motor neurons in human ALS and FALS. The highly penetrant dominant inheritance pattern of both the human and mouse diseases (4), together with the absence of motor neuron disease from “knockout” mice lacking endogenous murine SOD1 (5), strongly suggests that FALS pathology is because of a toxic gain-of-function in SOD. The biochemical nature of this toxic gain of function, however, and the mechanism by which SOD mutations cause the degeneration of motor neurons have remained elusive, largely because of the failure to identify novel properties of mutant SOD that are unambiguously linked to early cytopathological changes.
One hypothesis argues that toxicity results from the tendency of mutant SOD to “aggregate” into cytoplasmic inclusion bodies (6) that are evident in motor neurons from SOD transgenic mice (7, 8) and in cultured COS cells (9) or motor neurons (10) expressing mutant SOD cDNA. Cytoplasmic inclusion bodies are a hallmark of motor neuron degeneration in ALS and, indeed, of nearly all neurodegenerative diseases (11). SOD is itself a component of inclusion bodies in degenerating spinal cords from FALS patients (12, 13) and in end-stage mice expressing human FALS-linked SOD1 transgenes (6, 7, 12). How these inclusion bodies could cause neuronal degeneration—and indeed whether the inclusions are cytotoxic or even, perhaps, cytoprotective—is controversial. It has been suggested that neurotoxicity of mutant SOD arises from its possible coaggregation with and possible depletion of unidentified essential cellular components (6), although evidence in support of this hypothesis is lacking. Moreover, this model is inconsistent with biochemical studies indicating that protein aggregation occurs by specific interactions between folding intermediates and not by nonselective trapping (14). Moreover, formation of SOD inclusion bodies cannot contribute significantly to early FALS pathogenesis, because the appearance of detectable inclusion bodies in SOD1 transgenic mice is a late event—coinciding with the onset of overt motor neuron disease and the nearly synchronous loss of motor neurons (13, 15).
In the present study, we have investigated the pathway by which mutant SOD becomes incorporated into cytoplasmic inclusion bodies and the timing of this deposition relative to disease progression, in mice expressing human SOD1 transgenes. Our data show that aggregation—as defined by the formation of high molecular weight, insoluble protein complexes (IPCs)—and sequestration of these IPCs into cytoplasmic inclusion bodies—are separate and distinct processes. These data establish that SOD aggregation is indeed a property of FALS-linked mutations and that sequestration of SOD aggregates into intermediate filament-rich inclusion bodies occurs by means of retrograde transport on microtubules. Finally, we demonstrate that SOD IPCs are present in spinal cords from transgenic mice expressing human mutant but not wild-type SOD as early as postpartum day 30 (P30), at least 3 mo before either SOD inclusion bodies or overt motor neuron dysfunction are first manifested. The level of mutant SOD IPC in spinal cord increases steadily as the mice age. These data identify a progressive biochemical marker for FALS that is directly linked to a gain-of-function mutation, and suggest that aggregation of SOD into IPCs and not sequestration into inclusion body formation is an early event in the pathogenic mechanism.
Materials and Methods
Human embryonic kidney (HEK) cells were maintained and transfected as described (16). Human SOD cDNA (American Type Culture Collection, cat. no. 39786) served as a template for PCR amplification and mutagenesis to incorporate a C-terminal hemagglutinin (HA) epitope (YPYDVPDYA). This clone was inserted into pCDNA3 vector (Invitrogen) and mutated to produce the G85R and the G93A mutants. The sequences of all clones used in this study were verified on both strands by sequencing. We generated a rabbit polyclonal antibody, B1, to a peptide from human SOD (CESNGPVKVWGSIK) (7) conjugated by the N terminus to keyhole limpet hemocyanin. B1 antibody was affinity purified against the immunogen peptide. Additional polyclonal antibodies against SOD used in this study were from BioDesign (Kennebunk, ME; W59191R) and Calbiochem (574597). Anti-cystic fibrosis transmembrane conductance regulator (CFTR) polyclonal antibody has been described (17). Monoclonal antibodies used in this study were against vimentin (Sigma, V-6630), phosphorylated neurofilament (mixture of NF-M, NF-L, and NF-H subunits) (Sternberger–Meyer, Jarrettsville, MD; SMI 312), SOD (Sigma, S-2147), and HA (Covance MMS-101P, Richmond, CA). Proteasome inhibitors N-acetyl-leucyl-leucyl-norleucinal (ALLN), MG132, and lactacystin were from Calbiochem. Z-L3VS was a kind gift of M. Bogyo and H. Ploegh, Harvard Medical School, Boston.
Transgenic mice expressing human wild-type and G93A mutant SOD (18) were obtained from The Jackson Laboratory [TgN(SOD10G93A)1Gur and TgN(SOD1)2Gur] and maintained as hemizygotes by mating transgenic males with B6SJLF1 females.
Immunofluorescence Microscopy.
HEK 293 cells were fixed and prepared for immunofluorescence as described (16) and viewed with a Zeiss Axiovert microscope by using a ×63, 1.4 NA objective, digitally acquired by using a cooled charge-coupled device camera (Princeton Instruments, Trenton, NJ) and Metamorph (Universal Imaging, Media, PA) software.
Spinal cords sections were prepared for immunohistochemistry (7) by using affinity-purified B1 anti-SOD polyclonal antibody and Texas red-X secondary antibody (Molecular Probes) and with mAb SMI312 and Alexa-488 secondary antibody (Molecular Probes) and imaged with a ×63 1.4 NA objective (Zeiss) and an MRC1000 confocal microscope (Bio-Rad). All micrographic images were processed with National Institutes of Health image software and prepared by using photoshop 4.0 (Adobe Systems, Mountain View, CA).
Pulse–Chase Analysis, Immunoprecipitation, and Immunoblotting.
Pulse–chase analysis of SOD-transfected HEK 293 cells was performed as described (17), except that pulse labeling was with 35S[Cys] for 45 min. Labeled cells were washed once in PBS, chased in complete medium, immunoprecipitated with monoclonal anti-HA antibody, separated on 4–20% SDS/PAGE, and analyzed by phosphorimaging (Molecular Dynamics). Immunoblotting of cell lysates from transfected HEK 293 cells expressing SOD was as described (16). Spinal cord extracts were prepared as described (7).
Results
Stability and Aggregation of Mutant FALS-Linked SOD.
The steady-state level of two FALS-linked SOD mutants, G93A and G85R, expressed in HEK 293 cells (Fig. 1A) was consistently lower than that of wild-type SOD expressed under identical conditions, suggesting that these mutations may interfere with either the folding efficiency or the stability of SOD. Overnight treatment with proteasome inhibitors increased the steady-state levels of the 22-kDa monomeric form of mutant SOD but had no significant effect on the level of the wild-type protein, further implying that the reduced levels of G85R and G93A may be because of increased turnover by the proteasome. Although SOD was completely soluble in nonionic detergent and migrated exclusively as monomer in the presence or absence of proteasome inhibitor, some G93A and G85R, exhibited significantly reduced mobility, producing an immunoreactive smear that extended through the gel into the sample wells. We consistently observed in proteasome inhibited cells expressing either G93A or G85R, and to a lesser extent in untreated cells, three prominent HA-SOD immunoreactive electrophoretic species with apparent molecular masses of 22, 44, and 66 kDa—1-, 2-, and 3-fold, respectively, the apparent molecular mass of monomeric SOD (asterisks), suggesting that the mutant proteins may form nonnative, SDS-stable homooligomers.
Figure 1.

IPC formation by mutant SOD. (A) Effect of proteasome inhibition on electrophoretic mobility and detergent solubility of SOD. HEK cells transiently expressing wild-type SOD (lanes 1–4 and 9–12), G85R (lanes 5–8), or G93A (lanes 13–16) mutant SOD were treated overnight with proteasome inhibitor (ALLN, 10 μg/ml), lysed, and separated into nonionic detergent soluble (s) and insoluble (i) fractions and analyzed on immunoblots probed with a mAb to the HA epitope tag. Mobility of molecular weight markers in kDa is indicated at Left. Asterisks denote positions of high molecular SOD at ×2 and ×3 the apparent molecular weight of monomeric SOD. (B) Decreased stability of mutant SOD. HEK cells transiently expressing wild-type (○ and □) or G85R (● and ■) SOD were pulse-labeled with [35S]Cys for 45 min and chased for the times indicated in the presence (□ and ■) or absence (○ and ●) of the proteasome inhibitor lactacystin (5 μM). Data are mean ± SEM from 4–6 independent experiments. Similar data were obtained for the G93A mutant.
We used pulse–chase analysis to evaluate the stability of mutant and wild-type HA-tagged SOD expressed in HEK cells (Fig. 1B). Wild-type SOD was extremely stable (t1/2 > 24 h) consistent with previous reports (19, 20); its stability was unaffected by treatment of the cells with proteasome inhibitor. By contrast, G85R (Fig. 1B) and G39A (data not shown) were significantly less stable (t1/2 ≈ 7 h); this decay was blocked by the proteasome inhibitors lactacystin (Fig. 1B).
The steady-state level of most cytoplasmic proteins is maintained by the dynamic balance between protein synthesis and proteasome-dependent proteolysis; the above data suggest that the reduced levels of mutant relative to wild-type SOD in transfected HEK cells is the result of increased degradation. If prevented from degradation, mutant SOD forms high molecular weight aggregates that are partially insoluble in both nonionic detergent (judged by partitioning) and SDS (judged by the extensive smear on the gel). We call these aggregated forms IPCs. IPC formation cannot be an artifact of exposure to proteasome inhibitors, because some high molecular weight aggregates of mutant SOD are routinely present in transfected cells even in the absence of proteasome inhibitors (Fig. 1A) and in the spinal cords of G93A transgenic mice (see later). These data demonstrate that the reduced half-life and the tendency to form IPCs are gains-of-function intrinsic to these FALS-linked SOD mutants.
Mutant SOD Is Delivered to Aggresomes by a Microtubule-Dependent Process.
Aggresomes are pericentriolar inclusion bodies into which aggregated proteins are delivered by retrograde transport on microtubules (16). To determine whether aggregated FALS-linked SOD mutants are targeted to aggresomes, we examined the intracellular distribution of wild-type and mutant SOD in HEK cells by indirect immunofluorescence microscopy (Fig. 2A). Wild-type and mutant SOD were diffusely distributed in both nuclear and cytoplasmic compartments in the absence of proteasome inhibitor. Exposure to lactacystin had no effect on the distribution of wild-type SOD, but induced redistribution of a fraction of G85R and G93A into discrete, juxtanuclear structures reminiscent of the “spheroids” that characterize degenerating motor neurons in ALS. These structures were rarely (<1% of cells, Fig. 2B) observed in cells expressing wild-type SOD, irrespective of the presence of proteasome inhibitor. In marked contrast, a significant number of cells expressing either mutant contained prominent SOD-positive inclusions (Fig. 2B); this fraction was increased ≈5-fold by exposure to lactacystin.
Figure 2.

Mutant SOD accumulates in intracellular inclusion bodies. (A) HEK cells were transfected wild-type or mutant SOD as indicated. Cells on the Right (c, d, g, h, k, and l) were treated overnight with lactacystin, followed by examination by indirect immunofluorescence with an affinity-purified antibody to SOD (α-SOD; a, c, e, g, i, and k) and a mAb to the HA epitope tag (α-SOD; b, d, f, h, j, and l). (Bar = 15 μm.) (B) Cells transfected and treated as in A were counted and scored for the presence of inclusion bodies. Between 200 and 430 cells were counted for each condition. Single (P < 0.05) and double (P < 0.001) asterisks indicate statistical significance relative to untreated SOD based on Student's t test.
To determine whether the inclusion bodies observed in HEK cells expressing mutant SOD are aggresomes, we compared the distribution of wild-type and mutant SOD with that of several aggresomal markers (Fig. 3). Both G85R (Fig. 3A) and G93A (not shown) inclusions strongly and consistently colocalized with GFP-CFTR, which we have previously shown to be sequestered into aggresomes (16). By contrast, no colocalization was observed between aggresomal GFP-CFTR and wild-type SOD (not shown). Moreover, G85R (Fig. 3B) and G93A (data not shown), but not wild-type (Fig. 3B) SOD, were colocalized with the centrosomal marker pericentrin, suggesting that SOD, like other aggresomally targeted proteins (16, 21), accumulates at the microtubule organizing center. To evaluate the role of the microtubule cytoskeleton in the formation of mutant SOD inclusion bodies, cells expressing G85R were treated with proteasome inhibitor in the absence or presence of nocodazole, a drug that induces spontaneous depolymerization of microtubules (Fig. 3C). As previously demonstrated for aggresomes formed from CFTR (16) and other proteins (21), nocodazole completely abrogated the redistribution of G85R into aggresomes, demonstrating that delivery of mutant SOD to inclusion bodies in HEK cells requires an intact microtubule cytoskeleton.
Figure 3.

Inclusion bodies containing mutant SOD are aggresomes. (A) Colocalization of G93A SOD with ΔF508 CFTR aggresomes. HEK cells were cotransfected with G93A and ΔF508 CFTR, treated with MG132 before examination by double indirect immunofluorescence for CFTR (α-CFTR; b) and HA epitope tag (α-HA; a). (B) Colocalization of SOD and pericentrin. HEK cells were transfected with wild-type (a–c) or G85R mutant (d–f) SOD, examined by double indirect immunofluorescence by using a polyclonal antibody to pericentrin (α-PC; b and e) and mAb to the HA epitope tag (α-HA; a and d). (C) Delivery of SOD to aggresomes requires an intact microtubule cytoskeleton. HEK cells expressing G85R SOD were exposed to proteasome inhibitor (ALLN, 10 μg/ml) in the presence or absence of nocodazole (10 μg/ml) as indicated. SOD immunolocalization was detected with α-HA mAb. (D) Vimentin colocalizes with mutant but not wild-type SOD. HEK cells expressing wild-type (a–c) or G93A (d–f) SOD were exposed overnight to the proteasome inhibitor MG132, examined by double indirect immunofluorescence by using a polyclonal antibody to SOD (α-SOD; a and c) and mAb to vimentin (α-vim; b and e). (Bar = 15 μm.) (E) Vimentin collapses around SOD aggresomes in the absence of proteasome inhibitor. HEK cells expressing wild-type (a–c) or G93A (d–f) SOD were examined by immunofluorescence with antibodies aginat SOD (a and c) and (b and e). (Bar = 15 μm.)
A hallmark of aggresomes (16) and of many neurodegeneration-associated intracellular inclusion bodies (11), including those found in ALS (22, 23), is the deposition of abnormal intermediate filament (IF) proteins—vimentin in HEK cell aggresomes and neurofilament in neuronal inclusions (22, 23). We found that vimentin was collapsed around SOD-immunopositive aggresomes in proteasome-inhibited HEK cells expressing G85R (Fig. 3D) and G93A (data not shown); a similar distribution of vimentin was observed to encircle spontaneously formed aggresomes from HEK cells expressing mutant SOD in the absence of proteasome inhibitor (Fig. 3E). By contrast, vimentin was distributed in a characteristic extended fibrous distribution in cells expressing similar levels of wild-type SOD in the absence, but not the presence, of proteasome inhibitor, consistent with our previous finding that maintenance of proper interphase distribution of intermediate filaments requires active proteasomes (16).
SOD IPCs and Inclusion Bodies in Motor Neurons from G93A Transgenic Mice.
Mice expressing the G93A transgene exhibit normal motor function for the first 3–4 mo of age. They become paralyzed in one or more limbs by 4–5 mo of age, and progress within 2 wk to exhibit a severe pathology characterized initially by extensive vacuolation of the cytoplasm of large motor neurons of the anterior horn of the spinal cord (18, 24) and disruption of the Golgi apparatus (25) and, in end-stage animals, by severe depletion of anterior motor neurons. Surviving motor neurons exhibit hyaline filamentous, neurofilament-rich, spheroidal inclusions indistinguishable from the pathological structures present in spinal motor neurons in the human disease, whereas mice expressing similar or higher levels of wild-type human SOD do not develop motor neuron disease or spinal pathology (18). We used confocal microscopy to examine the intracellular distribution of SOD and neurofilament in sections of spinal cord from a well-characterized line of transgenic mice that expresses human G93A SOD and a control line that expresses comparable levels of human wild-type SOD (ref. 18); (Fig. 4).
Figure 4.
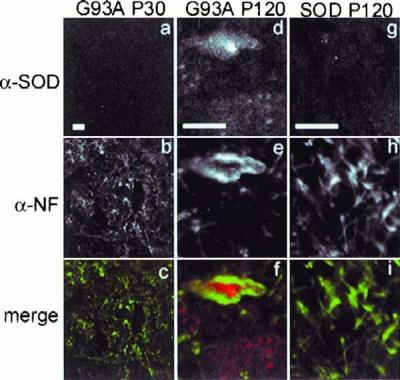
Inclusion bodies in spinal cords from G93A transgenic mice. Abnormal neurofilament deposition around SOD inclusions in spinal cord motoneurons in G93A transgenic mice at P120 (d–f) but not at P30 (a–c). Morphology of P30 motoneurons is indistinguishable from that of P120 SOD mice (g–i) and from nontransgenic mice (not shown). Spinal cord sections from mice expressing human SOD transgenes were double labeled by indirect immunofluorescence using affinity purified polyclonal antibody to SOD (a, d, and g) and mAb to phosphorylated neurofilament (b, e, and h) and visualized by confocal microscopy. Note magnification in a–d is lower than that in d–i. (Bars = 10 μm.)
Spinal cords from P30 G93A (Fig. 4) or P120 control mice expressing similar levels of SOD (see Fig. 5) and nontransgenic mice (data not shown) exhibited weak, diffuse immunoreactivity and normal neurofilament distribution that was largely restricted to axonal processes. By contrast, intense SOD immunoreactivity was routinely observed in large (>10 μm) cytoplasmic inclusions in spinal cord motor neurons from P120 mice expressing G93A SOD together with severely deranged neurofilament, which were mostly present in soma where they appear to encircle the G93A inclusions reminiscent of the distribution of vimentin around HEK cell SOD aggresomes.
Figure 5.

Accumulation of SOD in IPCs is an early and specific event in spinal cord from mice expressing G93A human SOD. (A) Expression of SOD in transgenic and control mice. Immunoblots of spinal cord extracts (equal μg total protein per lane) from two different mice expressing a wild-type human SOD transgene (lanes 1 and 3), a G93A human SOD transgene at P30 (lane 4) and P120 (lane 5), and from a nontransgenic littermate (lane 2) probed with affinity-purified B1 polyclonal SOD antibody. Blots on Right and Left differ only in the length of time of exposure of the chemilluminescence reaction to film. (B) SOD IPCs increase with age in G93A spinal cords. An immunoblot similar to that in A was probed with affinity-purified B1 antibody. Intensity of high molecular weight, B1-immunoreactive material was quantified from a digital scan of the blot by integrating the total pixel density after subtraction of background intensity from a nontransgenic control lane.
Immunoblot analysis of SOD expression in spinal cord extracts from P30 and P120 mice (Fig. 5A) revealed a single band corresponding to the mobility of monomeric SOD (≈22 kDa) in P120 mice expressing wild-type SOD and in P30 or P120 mice expressing the G93A transgene, but not in nontransgenic animals, confirming that all of the transgenic mice expressed comparable levels of the transgenic protein. Remarkably, when the same blot was exposed to film for a longer time (Fig. 5 Right), higher molecular weight forms of SOD-immunoreactive protein were evident—exclusively in samples from the G93A spinal cords. Identical results were obtained from several different animals by using different anti-SOD antibodies, but not with irrelevant or preimmune antisera, confirming that the high molecular weight forms are composed of human SOD. Intermediate levels of aggregated SOD were observed in spinal cords from P60 and P90 mice (Fig. 5B), indicating that the total amount of SOD in IPCs increases continuously as the animals age. As observed in HEK cells expressing mutant SOD, the high molecular mass material consisted of prominent bands at 44, 66, and 88 kDa and an additional higher molecular mass smear that ran to the top of the gel, indicating that some of the mutant SOD was present in IPCs similar to those observed in HEK cells. Together, these data establish that G93A SOD is present in spinal cord IPCs even from young mice that lack microscopic SOD inclusion bodies and disease pathology.
Discussion
The data in this paper demonstrate that FALS mutations promote SOD conversion to IPCs and thus define a biochemical marker corresponding to a mutationally linked SOD gain-of-function. Previous reports of SOD “aggregation” in FALS have relied on the identification of microscopically visible inclusion bodies—which, in transgenic mice, are detectable only once disease is evident. Our data demonstrate that SOD IPCs are present at P30—a time before the manifestation of ALS pathology, and several months before the appearance of inclusion bodies (6, 7, 24, 26, 27). These findings, which reveal that aggregation of SOD into IPCs and inclusion body formation occur in spinal cord with distinctly different time courses, have significant implications for understanding the role of SOD aggregation in FALS pathogenesis.
Previous studies have used immunolocalization of SOD to inclusion bodies to assess SOD “aggregation” in mice (6) and in cell culture models (9, 10). Our data indicate that aggregated SOD can be differentiated from the native protein on the basis of detergent solubility and electrophoretic mobility. We term this nonnative SOD form an insoluble protein complex (IPC) to more precisely reflect its physical state and to circumvent the use of the term “aggregate,” which to some investigators means “inclusion body.” In experiments (not shown) by using coexpression of mutant SOD and His6, myc-tagged ubiquitin (28) we could detect no significant level of ubiquitination, suggesting that the reduced mobility of SOD immunoreactive material was not because of extensive modification with ubiquitin. Similarly, only weak ubiquitin immunoreactivity was detected (data not shown) in the intracellular aggresome-like inclusion bodies (see below) into which the mutant SOD was sequestered. These data do not address the probable role of ubiquitin in targeting mutant, unaggregated SOD for degradation by the proteasome. They indicate, however, that the decrease in SOD mobility associated with IPCs is because of aggregation into complexes, not extensive ubiquitination.
We show here that G93A and G85R SOD in transfected cells is delivered to aggresomes—vimentin-rich inclusion bodies—by a microtubule-dependent process. A similar mechanism may underlie the deposition of mutant SOD into NF-encased inclusion bodies in spinal motor neurons in FALS and in FALS transgenic mice. Although the process of aggregation—namely the conversion of misfolded protein monomers into high molecular weight IPCs—is likely to be driven by a process of diffusion-limited seeded protein polymerization (29), the sequestration of mutant SOD into inclusion bodies appears to require their active transport on microtubules (16). Because the solubility of mutant SOD is unaffected by treatment of cells with nocodazole (J.A.J. and R.R.K., unpublished data), it is likely that IPC formation does not require sequestration of mutant SOD inclusion bodies. It is likely, therefore, that IPCs are the form of SOD that is subject to retrograde transport on microtubules. This is reasonable because the large molecular dimensions of high molecular weight IPCs, and their often extended shape, would severely impede diffusive movement through the cell. We have termed these types of inclusion bodies aggresomes, to emphasize the fact that they are formed by a specific, energy requiring, dynein-mediated process directed toward the minus end of microtubules (16). Although originally shown for dislocated integral membrane proteins (16), it is evident from the present work and from recently published data (21) that aggresomes can also form from misfolded variants of cytosolic proteins. As it is not possible to disrupt MT structure or transport in SOD transgenic mice in vivo, we are not able at present to directly test the hypothesis that ALS-linked SOD inclusions in motor neurons are formed by an active MT-based process. Moreover, because SOD-NF inclusions in FALS are often present in axons, and because of the unique discontinuous structure of axonal microtubules (31, 32), not all SOD-positive motor neuron inclusions will be expected to colocalize with centrosomal markers. Nonetheless, the data presented in this paper establish that sequestration of aggregated SOD into inclusion bodies in nonneuronal cells is a MT-dependent process and suggest that a similar mechanism exists in neurons.
The early appearance of SOD IPCs in spinal cord from transgenic mice suggests that IPC formation may contribute to the toxic gain-of-function that underlies the pathogenesis of FALS. Because these aggregates are likely substrates for dynein-mediated retrograde transport on microtubules, it is possible that the presence of an increasing burden of IPCs could disrupt microtubule-dependent axonal transport of other substrates, some of which may be essential for neuronal viability. It is noteworthy that disruption of both fast (33) and slow (34) components of axonal transport are among the earliest detectable cellular abnormalities in motor neurons from ALS transgenic mice. Competition by aggregated SOD could disrupt other retrograde substrates such as Golgi cisternae, which are maintained in a pericentriolar distribution by active transport on MT (35), and could thus explain the disruption of Golgi structure that is observed in G93A transgenic mice (25) and in human ALS (36). Additional studies will be required to assess how the interaction of mutant proteins like G93A or G85R SOD with the microtubule transport apparatus might affect these crucial transport processes.
Although the data presented here demonstrate that SOD aggregation into IPCs is an early event in FALS pathogenesis, they do not exclude the possibility that abnormal oxidative mechanisms may also contribute to the neurotoxic gain-of-function that underlies this disease. Mutant SOD can catalyze enhanced production of toxic free radicals (37) and reactive oxygen species such as peroxynitrite (38) in vitro and is associated with increased oxidative protein damage and increased tissue content of reactive oxygen species in G93A mice in vivo (39). On the other hand, aggregated proteins that the cell is unable to efficiently clear are likely to accumulate oxidative damage.
Oxidative free radicals and reactive oxygen species can damage proteins by modification of amino acid side chains, fragmentation of polypeptide chains, and induction of both covalent and noncovalent aggregation; all of these adducts are substrates for proteasome-mediated degradation (40). The production of an abundant misfolded protein like SOD, compounded by the generation of oxidatively damaged proteins imposes an increased burden on the ubiquitin-proteasome pathway. The steady-state presence of SOD IPCs in spinal cords from mice expressing the G93A transgene indicates that the capacity of this disposal system to eliminate misfolded SOD must be exceeded even at P30. We propose that the accumulation of SOD IPCs could cause neuron death by eventually overwhelming the capacity of the ubiquitin proteasome pathway to degrade, not only malfolded SOD molecules and oxidatively damaged proteins, but in the end, critical regulatory factors that are known substrates of the proteasome, such as cyclins, cyclin-dependent kinases and transcription factors. Our observation that proteasome inhibitors amplify and accelerate the degenerative process in cultured cells, together with recent evidence suggesting that dysfunction of the ubiquitin pathway can directly lead to neurodegenerative disease (30, 41–43), supports our hypothesis. Further studies will clearly be required to clarify the relationships among IPC formation, the ubiquitin proteasome pathway, and the pathogenesis of neurodegenerative disease.
Acknowledgments
We thank J. Frydman and members of the Kopito lab for helpful discussions, G. Kaung and Tim Fleck for technical assistance, and J. McLaughlin, R. Phillips, and S. Brooke for help with histology. This work was supported by grants from the National Institutes of Health. M.D. was supported by a National Science Foundation predoctoral training grant.
Abbreviations
- ALLN
N-acetyl-leucyl-leucyl-norleucinal
- ALS
amyotrophic lateral sclerosis
- FALS
familial ALS
- HEK
human embryonic kidney 293
- IPC
insoluble protein complex
- Pn
n days postpartum
- SOD
superoxide dismutase
- CFTR
cystic fibrosis transmembrane conductance regulator
- HA
hemagglutinin
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.220417997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.220417997
References
- 1.Siddique T. Adv Neurol. 1991;56:227–231. [PubMed] [Google Scholar]
- 2.Emery A, Holloway S. In: Human Motor Neuron Diseases. Rowland L, editor. New York: Raven; 1982. pp. 139–147. [Google Scholar]
- 3.Rosen D R, Siddique T, Patterson D, Figlewicz D A, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan J P, Deng H X, et al. Nature (London) 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 4.Siddique T, Nijhawan D, Hentati A. Neurology. 1996;47:S27–S35. doi: 10.1212/wnl.47.4_suppl_2.27s. [DOI] [PubMed] [Google Scholar]
- 5.Reaume A G, Elliott J L, Hoffman E K, Kowall N W, Ferrante R J, Siwek D F, Wilcox H M, Flood D G, Beal M F, Brown R H, Jr, et al. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 6.Bruijn L I, Houseweart M K, Kato S, Anderson K L, Anderson S D, Ohama E, Reaume A G, Scott R W, Cleveland D W. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- 7.Bruijn L I, Becher M W, Lee M K, Anderson K L, Jenkins N A, Copeland N G, Sisodia S S, Rothstein J D, Borchelt D R, Price D L, Cleveland D W. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 8.Tu P H, Raju P, Robinson K A, Gurney M E, Trojanowski J Q, Lee V M. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koide T, Igarashi S, Kikugawa K, Nakano R, Inuzuka T, Yamada M, Takahashi H, Tsuji S. Neurosci Lett. 1998;257:29–32. doi: 10.1016/s0304-3940(98)00800-3. [DOI] [PubMed] [Google Scholar]
- 10.Durham H D, Roy J, Dong L, Figlewicz D A. J Neuropathol Exp Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Mayer R J, Lowe J, Lennox G, Doherty F, Landon M. Prog Clin Biol Res. 1989;317:809–818. [PubMed] [Google Scholar]
- 12.Shibata N, Hirano A, Kobayashi M, Siddique T, Deng H X, Hung W Y, Kato T, Asayama K. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 13.Shibata N, Kobayashi M, Hirano A, Asayama K, Horiuchi S, Dal Canto M, Gurney M E. Acta Hostochem Cytochem. 1999;32:17–30. [Google Scholar]
- 14.Speed M A, Wang D I, King J. Nat Biotechnol. 1996;14:1283–1287. doi: 10.1038/nbt1096-1283. [DOI] [PubMed] [Google Scholar]
- 15.Morrison B M, Morrison J H, Gordon J W. J Exp Zool. 1998;282:32–47. [PubMed] [Google Scholar]
- 16.Johnston J A, Ward C L, Kopito R R. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ward C L, Kopito R R. J Biol Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- 18.Gurney M E, Pu H, Chiu A Y, Dal Canto M C, Polchow C Y, Alexander D D, Caliendo J, Hentati A, Kwon Y W, Deng H X, et al. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 19.Hoffman E K, Wilcox H M, Scott R W, Siman R. J Neurol Sci. 1996;139:15–20. [PubMed] [Google Scholar]
- 20.Borchelt D R, Lee M K, Slunt H S, Guarnieri M, Xu Z S, Wong P C, Brown R H, Jr, Price D L, Sisodia S S, Cleveland D W. Proc Natl Acad Sci USA. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Mata R, Bebok Z, Sorscher E J, Sztul E S. J Cell Biol. 1999;146:1239–1254. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delisle M B, Carpenter S. J Neurol Sci. 1984;63:241–250. doi: 10.1016/0022-510x(84)90199-0. [DOI] [PubMed] [Google Scholar]
- 23.Hirano A. Adv Neurol. 1991;56:91–101. [PubMed] [Google Scholar]
- 24.Dal Canto M C, Gurney M E. Am J Pathol. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- 25.Mourelatos Z, Gonatas N K, Stieber A, Gurney M E, Dal Canto M C. Proc Natl Acad Sci USA. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ripps M E, Huntley G W, Hof P R, Morrison J H, Gordon J W. Proc Natl Acad Sci USA. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shibata N, Hirano A, Kobayashi M, Dal Canto M C, Gurney M E, Komori T, Umahara T, Asayama K. Acta Neuropathol. 1998;95:136–142. doi: 10.1007/s004010050777. [DOI] [PubMed] [Google Scholar]
- 28.Ward C L, Omura S, Kopito R R. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 29.Lansbury P T., Jr Neuron. 1997;19:1151–1154. doi: 10.1016/s0896-6273(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 30.van Leeuwen F W, de Kleijn D P, van den Hurk H H, Neubauer A, Sonnemans M A, Sluijs J A, Koycu S, Ramdjielal R D J, Salehi A, Martens G J M, et al. Science. 1998;279:242–247. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- 31.Bray D, Bunge M B. J Neurocytol. 1981;10:589–605. doi: 10.1007/BF01262592. [DOI] [PubMed] [Google Scholar]
- 32.Sharp G A, Osborn M, Weber K. J Cell Sci. 1981;47:1–24. doi: 10.1242/jcs.47.1.1. [DOI] [PubMed] [Google Scholar]
- 33.Zhang B, Tu P, Abtahian F, Trojanowski J Q, Lee V M. J Cell Biol. 1997;139:1307–1315. doi: 10.1083/jcb.139.5.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williamson T L, Cleveland D W. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]
- 35.Lippincott-Schwartz J. Curr Opin Cell Biol. 1998;10:52–59. doi: 10.1016/s0955-0674(98)80086-0. [DOI] [PubMed] [Google Scholar]
- 36.Mourelatos Z, Hirano A, Rosenquist A C, Gonatas N K. Am J Pathol. 1994;144:1288–1300. [PMC free article] [PubMed] [Google Scholar]
- 37.Yim M B, Kang J H, Yim H S, Kwak H S, Chock P B, Stadtman E R. Proc Natl Acad Sci USA. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crow J P, Sampson J B, Zhuang Y, Thompson J A, Beckman J S. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- 39.Andrus P K, Fleck T J, Gurney M E, Hall E D. J Neurochem. 1998;71:2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x. [DOI] [PubMed] [Google Scholar]
- 40.Grune T, Davies K J. Biofactors. 1997;6:165–172. doi: 10.1002/biof.5520060210. [DOI] [PubMed] [Google Scholar]
- 41.Saigoh K, Wang Y L, Suh J G, Yamanishi T, Sakai Y, Kiyosawa H, Harada T, Ichihara N, Wakana S, Kikuchi T, Wada K. Nat Genet. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- 42.Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein M J, Jonnalagada S, Chernova T, et al. Nature (London) 1998;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- 43.Fergusson J, Landon M, Lowe J, Ward L, van Leeuwen F W, Mayer R J. Neurosci Lett. 2000;279:69–72. doi: 10.1016/s0304-3940(99)00917-9. [DOI] [PubMed] [Google Scholar]