

Abstract
Chronic beryllium disease results from beryllium exposure in the workplace and is characterized by CD4+ T cell-mediated inflammation in the lung. Susceptibility to this disease is associated with particular HLA-DP alleles. We isolated beryllium-specific T cell lines from the lungs of affected patients. These CD4+ T cell lines specifically responded to beryllium in culture in the presence of antigen-presenting cells that expressed class II MHC molecules HLA-DR, -DQ, and -DP. The response to beryllium was nearly completely and selectively blocked by mAb to HLA-DP. Additional studies showed that only certain HLA-DP alleles allowed presentation of beryllium. Overall, the DP alleles that presented beryllium to disease-specific T cell lines match those implicated in disease susceptibility, providing a mechanism for this association. Based on amino acid residues shared by these restricting and susceptibility DP alleles, our results provide insight into the residues of the DP β-chain required for beryllium presentation.
Exposure to beryllium in the workplace continues to be a public health concern with approximately 800,000 individuals exposed and potentially at risk for developing chronic beryllium disease (CBD) (1). Of those exposed, CBD develops in up to 16% of workers, depending on characteristics of the exposure (2–4) and genetic susceptibility of the individual (5–7). The disease is characterized by granulomatous inflammation, primarily involving the lung (1). Considerable evidence indicates that CD4+ T cells are important in the immunopathogenesis of CBD, and the development of granulomatous inflammation in the lung is associated with the accumulation of CD4+ T cells in the bronchoalveolar lavage (BAL) (8–11). In the presence of antigen-presenting cells expressing class II MHC molecules, these T cells can be shown to proliferate and release cytokines in response to beryllium sulfate (BeSO4) in culture (10). How these beryllium-reactive CD4+ T cells interact with the beryllium/peptide/MHC complex and what allows for presentation of beryllium is currently unknown.
Susceptibility to CBD has been associated with particular alleles of the class II MHC molecule, HLA-DP (5–7, 12). In the present study, we determined whether proliferation of beryllium-specific CD4+ T cells from the lungs of CBD patients involved presentation by HLA-DP. We found that proliferation of each T cell line tested was nearly completely inhibited by the addition of anti-DP mAbs. Proliferation studies also showed that the restricting DP molecules and other DP molecules able to present beryllium in culture match those known to be associated with increased susceptibility to disease. In addition to providing evidence of how these DP alleles function to increase disease susceptibility, the results pinpoint residues of the DP β-chain required for beryllium presentation.
Materials and Methods
Study Population and HLA Typing.
The diagnosis of CBD was established by using previously defined criteria (9, 13), including a history of exposure to beryllium, the presence of granulomatous inflammation on lung biopsy, and a positive proliferative response of BAL T cells to BeSO4 in vitro. Informed consent was obtained from each CBD patient, and the protocol was approved by the Human Subject Institutional Review Board at the University of Colorado Health Sciences Center and National Jewish Medical and Research Center.
HLA typing was performed by standard molecular techniques at the University of Colorado Health Sciences Center Clinical Immunology Histocompatibility Laboratory. The class II HLA haplotypes of the CBD patients and control subjects used in this study are shown in Table 1. The DPA1 allele for CBD patients 1, 2, and 3 was determined to be DPA1*0103.
Table 1.
HLA DRB1, DQB1, and DPB1 alleles of CBD patients and controls studied
| Patient and control population | DRB1 | DQB1 | DPB1 |
|---|---|---|---|
| CBD patient 1 | 1101 | 0301 | 0201 |
| CBD patient 2 | 1301, 1501 | 0602, 0603 | 0201, 0401 |
| CBD patient 3 | 0701, 1301 | 0202, 0603 | 0402, 1001 |
| CBD patient 4 | 0701, 0802 | 0202, 0301 | 0201, 1701 |
| CBD patient 5 | 0701, 1101 | 0202, 0301 | 0401, 1701 |
| Control 1 | 1302, 1502 | 0601, 0603 | 0201, 0301 |
| Control 2 | 1302, 1502 | 0601, 0604 | 0201 |
| Control 3 | 1101, 1103 | 0301 | 0401, 0402 |
| Control 4 | 03, 1301 | 0201, 0603 | 0301, 0401 |
| Control 5 | 0401 | 0302 | 1001 |
| Control 6 | 0401 | 0302 | 0101, 0601 |
| Control 7 | 1501 | 0602 | 0401 |
| Control 8 | 1301 | 0603 | 0401 |
| Control 9 | 0401, 0404 | 0603 | 0401 |
| Control 10 | 0701 | 0202 | 0401 |
| Control 11 | 1301 | 0603 | 0101 |
| Control 12 | 0102, 0402 | 0501, 0302 | 0401, 0402 |
Preparation of Cells and Generation of Beryllium-Specific T Cell Lines.
Mononuclear cells from BAL and peripheral blood were prepared as described (11). Transformed B cell lymphoblastoid cell lines (LCLs) were prepared by infecting peripheral blood mononuclear cells (PBMCs) with supernatant from the Epstein–Barr virus-secreting marmoset cell line B95–8 in the presence of 0.5 μg/ml of cyclosporin A (Sandoz Pharmaceutical) as described (14). Beryllium-specific T cell lines were generated by stimulation of BAL cells in 24-well plates (Costar) with 2 ml of RPMI 1640 supplemented with 10% heat-inactivated FCS (HyClone), 20 mM Hepes, 1 mM sodium pyruvate, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine (all from Life Technologies, Gaithersburg, MD), and the addition of 1 × 10-5 M BeSO4. After 5 days of culture, cells were washed free of BeSO4, and the T cell lymphoblasts were expanded further in culture with media supplemented with 20 units/ml of human rIL-2 (R & D Systems). The T cell lines were maintained in culture by cycles of restimulation (every 2–3 weeks) with 1 × 10-5 M BeSO4 in the presence of autologous, irradiated LCL and further expanded in culture with rIL-2.
Lymphocyte Proliferation Assay.
T cell lines (1 × 105 cells) were cultured in 96-well flat-bottomed microtiter plates with 1 × 105 irradiated (9,000 rad) LCL cells or irradiated (3,000 rad) PBMCs in the presence of 1 × 10-5 M or 1 × 10-6 M BeSO4, 1 × 10-5 M Al2(SO4)3, or 5 μg/ml phytohemagglutinin. After 54 h of culture, the wells then were pulsed with 1 μCi of [3H]thymidine for an additional 18 h, and incorporation of radioactivity was determined by β-emission spectroscopy. Proliferation assays were performed in triplicate.
Inhibition of proliferation was attempted with mAbs to class II HLA molecules added at the beginning of culture. The T cell lines were cultured with LCL cells, 1 × 10-5 M BeSO4, and indicated amounts of anti-DR (hybridomas L243 and LB3.1, American Type Tissue Collection), anti-DP [hybridoma B7.21, a gift from Ian Trowbridge, Salk Institute, La Jolla, CA (15)], or anti-DQ [hybridoma SVPL3, a gift from Francesco Sinigaglia, Roche Milano Ricerche, Milan, Italy (16)]. Proliferation with and without the indicated mAb then was determined as described above, and the extent of inhibition was calculated.
In some experiments, the fibroblast cell line, DP8302, transfected with DPA1*0103 and DPB1*0201 (17), was used as antigen-presenting cells. The transfected fibroblast cells were fixed by resuspending pelleted cells in Hanks' balanced salt solution (Mediatech, Herndon, VA) with 2% paraformaldehyde for 10 min. The cells were again pelleted and resuspended in lysine wash solution for 20 min at room temperature and washed three times before use in culture.
Results
Development of BeSO4-Specific T Cell Lines from the Lungs of CBD Patients.
BAL cells from four patients with CBD were cultured in vitro in the presence of 1 × 10-5 M BeSO4. We previously have shown that this concentration of BeSO4 is optimal for T cell proliferation (11). After 5 days, the blasted T cells were isolated and expanded in the presence of rIL-2 for approximately 10 days. More than 95% of the blasted T cells expressed CD4. These cells were restimulated in the presence of BeSO4 and 20 units/ml rIL-2 by using autologous, irradiated Epstein–Barr virus-transformed LCL cells as antigen-presenting cells. After the second cycle of stimulation, the remaining T cells were tested for response to BeSO4 and to other metal antigens, such as Al2(SO4)3, in the presence of autologous, irradiated PBMCs or LCLs. A strong proliferative response was seen with the addition of BeSO4, whereas no proliferation was observed to Al2(SO4)3 (Fig. 1). In general, the beryllium-reactive CD4+ T cell lines used in the subsequent experiments were studied after two cycles of stimulation and were extremely polyclonal as determined by heterogeneous T cell receptor Vβ expression (data not shown).
Figure 1.

Stimulation of a representative T cell line derived from the BAL of CBD patient 1. Proliferation of the T cell line to phytohemagglutinin (PHA) (5 μg/ml), BeSO4 (1 × 10-5 M), and Al2(SO4)3 (1 × 10-5 M) in the presence of autologous, irradiated PBMCs as antigen-presenting cells is shown. The data are presented as the mean cpm ± SEM.
Inhibition of Responses to Beryllium with Anti-MHC Class II Antibodies.
The MHC class II molecules involved in the presentation of beryllium were determined by using mAbs to HLA-DR, -DQ, and -DP. A representative blocking experiment is shown in Fig. 2. With beryllium-specific T cell lines derived from CBD patient 1 and autologous LCL as antigen-presenting cells, the proliferative response to optimal concentrations of BeSO4 was reproducibly and completely inhibited with low concentrations of the anti-DP mAb, B7.21. In three experiments with cell lines from CBD patient 1, inhibition with anti-DP varied from 81% to 100%. With cell lines from CBD patient 2, inhibition with anti-DP was 82% and 96% in two experiments. Fig. 2 also shows the variable and partial inhibition observed with two anti-DR mAbs and the lack of inhibition with the anti-DQ mAb.
Figure 2.

Inhibition of the T cell proliferative responses to beryllium with mAbs to class II MHC molecules. T cell lines from CBD patient 1 were cultured with autologous LCL, 1 × 10-6 M BeSO4, and increasing concentrations of anti-class II mAb. Background proliferation without addition of BeSO4 was 6,262 ± 122, and stimulation to BeSO4 without inhibitory antibodies was 25,574 ± 739 cpm. The data are presented as percentage inhibition of stimulation as a function of the concentration of anti-class II mAbs.
To evaluate the specificity of the anti-DP reagent, blocking experiments were performed by using a T cell hybridoma (DR4hCII) specific for a peptide of human type II collagen and HLA-DR4 (18). In contrast to the complete inhibition with mAb to DR, the addition of the anti-DP mAb failed to inhibit the release of IL-2 after stimulation by peptide in the presence of DRB1*0401 expressing presenting cells (data not shown). These studies indicate that the anti-DP mediated inhibition of the proliferative response to beryllium is unrelated to any nonspecific effect on DR.
The near-complete inhibition of beryllium responses with anti-DP and partial inhibition with anti-DR was examined further by using a fibroblast line, DP8302, transfected to express DPA1*0103 and DPB1*0201 on its surface (17). This is the only class II molecule expressed by these cells, and it is the same DP molecule expressed by CBD patients 1, 2, and 4. Expression of only HLA-DP was confirmed by staining (data not shown). After pulsing with BeSO4 for 2 h, the fibroblast cells were capable of stimulating the beryllium-specific T cells lines (shown for CBD patient 2, Fig. 3). Perhaps related to the absence of important costimulatory molecules, presentation of beryllium with fibroblast cells was suboptimal compared with presentation with autologous LCL cells. Similar to studies described above, addition of low concentrations of the anti-DP mAb reduced proliferation to background levels (Fig. 3). In contrast, no inhibition of proliferation was seen by using the anti-DR mAb even at 30-fold higher concentrations. This failure to inhibit suggests that the low-level anti-DR blocking described above is mediated through specific binding to DR molecules expressed on the LCL, but not on the transfected fibroblasts.
Figure 3.

Stimulation of the T cell line from CBD patient 2 with a fibroblast cell line (DP8302) transfected to express only HLA-DP (DPB1*0201 and DPA1*0103). Stimulation with BeSO4 (1 × 10-5 M) is shown on the left. Inhibition of this response with the addition of anti-DR or anti-DP mAb is shown on the right, and data are presented for both absolute cpm ± SEM and percent inhibition.
HLA-DPB1 Versus DRB1 and DQB1 Allele Restriction of BeSO4-Specific T Cell Lines.
The restricting HLA-DPB1 and DRB1 alleles possibly involved in beryllium presentation for CBD patients 1–4 were determined by using a panel of allogeneic Epstein–Barr virus-transformed LCLs and PBMCs as antigen-presenting cells. CBD patient 1 is homozygous for DPB1*0201, DRB1*1101, and DQB1*0301. T cell lines from this patient responded to beryllium in the presence of autologous LCL or allogeneic cells that expressed DPB1*0201 (Fig. 4a). Although control 1 expresses both DPB1*0201 and DPB1*0301, this latter allele and other nonmatched DP alleles tested did not allow for beryllium presentation (Fig. 4a). Presenting cells matched with the responder cells for HLA-DR and DQ, but not DP, were unable to induce beryllium-specific proliferation of the T cell lines (Fig. 4a). Together, the data indicate that recognition of beryllium in the lung of CBD patient 1 is restricted by DPB1*0201 and not by DR or DQ.
Figure 4.

Determination of the class II MHC allele restriction of BeSO4-specific T cell lines from (a) CBD patient 1 and (b) CBD patient 2. Stimulation of T cell lines was performed with autologous LCLs or allogeneic antigen-presenting cells in the presence of 1 × 10-5 M or 1 × 10-6 M BeSO4. The stimulation index for each of the various presenting cells is shown over each bar, and the molecular typing of the DPB1 and DRB1 alleles for each antigen-presenting cell is shown on the right. Data are expressed as the mean cpm ± SEM.
Beryllium presentation in CBD patient 2 required consideration of two DPB1 alleles, *0201 and *0401. However, Fig. 4b shows that restriction of these T cell lines was limited to *0201. Thus, presenting cells from three DPB1*0201 individuals induced a vigorous proliferative response after the addition of BeSO4. Two of these had DR alleles unrelated to those in CBD patient 2. In contrast, presenting cells from two individuals with DPB1*0401 did not present beryllium. Presenting cells from control 8 were matched for DRB1*1301 (and DQB1*0603) with the responding cells, but also were unable to stimulate the responding T cells after the addition of BeSO4. Interestingly, stimulation was apparent with presenting cells from control 7, and by exclusion, DRB1*1501 shared between stimulator and responder seems likely to be the presenting HLA molecule in these cultures.
We also developed T cell lines from individuals with CBD who possessed DPB1 alleles separate from *0201 and *0401 (see Table 1). With responder cells from CBD patient 3, proliferation assays showed that antigen-presenting cells matched at either DPB1*0402 or DPB1*1001, and without any matching DR alleles, stimulated after addition of BeSO4 (data not shown). In contrast, antigen-presenting cells from separate individuals matched for DR or DQ alleles did not allow for presentation of beryllium. T cell lines derived from CBD patient 4 showed a similar restriction pattern in that both DP alleles (DPB1*0201 and DPB1*1701) allowed presentation of BeSO4 (data not shown). The only matching DR allele that could be tested was DRB1*0701, which did not present beryllium.
Ability of Additional Nonrestricting DPB1 Alleles to Stimulate BeSO4-Specific T Cell Lines.
While studying responses of the different T cell lines, we noted that antigen-presenting cells from some individuals mediated BeSO4-specific responses although they were not matched for either DP or DR alleles with the responder. For example, DPB1*0201 expressing LCLs were able to present beryllium to T cell lines from CBD patient 3 (DPB1*0402, DPB1*1001) (data not shown). DR molecules could be excluded because they were shown not to be able to present beryllium when expressed by presenting cells from other individuals who had the nonpresenting allele, DPB1*0401.
We used T cell stimulation in conjunction with anti-DP inhibition to implicate additional DP alleles capable of presenting beryllium. For example, presenting cells from three additional individuals mediated proliferation of the T cell lines from CBD patient 2 in the presence of BeSO4 (Fig. 5). The degree of proliferation was considerably less than that with the restricting DPB1*0201 allele (see Fig. 4b). To confirm that the low-level stimulation was because of beryllium presentation by DPB1 alleles, we also tested for anti-DP inhibition. As shown in Fig. 5, beryllium-specific proliferation was completely inhibited by the anti-DP mAb. These and other similar studies indicated that additional DPB1 alleles, such as *0402, *0601, and *1001, are capable of presenting BeSO4.
Figure 5.

Stimulation of the T cell line from CBD patient 2 using nonrestricting DPB1 alleles. In the presence of 1 × 10-5 M BeSO4, the T cell line from CBD patient 2 was stimulated with various antigen-presenting cells not matched for this individual's DP or DR alleles. Stimulation also was inhibited by the addition of 10 μg/ml of anti-DP mAb. The stimulation indices for each of the various conditions and the molecular typing of the DPB1 and DRB1 alleles for each antigen-presenting cell are shown. Data are expressed as the mean cpm ± SEM.
DP β-chain Sequences of Beryllium-Presenting and Nonpresenting Alleles.
Table 2 shows the polymorphic amino acid residues of the HLA-DP β-chains that we studied for their ability to present beryllium. DPB1*0101 and DPB1*0401 alleles were repeatedly unable to stimulate the T cell lines of any of the four CBD patients after addition of BeSO4 in contrast to the other alleles shown. The presenting alleles show major differences in sequence at position 55–56 and a conservative change at position 36. Of note, except for DPB1*0402, all beryllium-presenting alleles have a glutamate (E) at position 69 compared with neither of the nonpresenting alleles.
Table 2.
DP β-chain sequences of beryllium-presenting and nonpresenting alleles
| Ability to present Be | Amino acid position
|
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 8 | 9 | 11 | 17 | 33 | 35 | 36 | 55 | 56 | 57 | 65 | 68 | 69 | 76 | 84 | 85 | 86 | 87 | ||
| DPB1*0101 | Neg | Y | V | Y | G | A | E | Y | A | A | A | E | I | E | K | V | D | E | A | V |
| DPB1*0401 | Neg | – | L | F | – | – | – | F | – | – | – | – | – | – | – | M | G | G | P | M |
| DPB1*0201 | + | – | L | F | – | – | – | F | V | D | E | – | – | – | E | M | G | G | P | M |
| DPB1*0402 | + | – | L | F | – | – | – | F | V | D | E | – | – | – | – | M | G | G | P | M |
| DPB1*0601 | + | – | – | – | L | – | – | F | V | D | E | D | L | – | E | M | – | – | – | – |
| DPB1*1001 | + | – | – | H | L | – | – | F | V | D | E | – | – | – | E | – | – | – | – | – |
| DPB1*1701 | + | – | – | H | L | – | – | F | V | D | E | D | – | – | E | M | – | – | – | – |
Sequences for the different alleles are presented based on homology to DPB1*0101, and a dash indicates the same amino acid at that position. The bolded amino acid residues indicate charged sequence differences in presenting alleles compared to DPB1*0101.
Discussion
Little is currently known regarding the mechanism of beryllium sensitization and why only a minority of exposed individuals develop chronic inflammatory lung disease. In addition to the type of exposure, genetic susceptibility appears to be an important variable, and particular HLA-DP alleles have been associated with the development of this disease (5–7). The present study addresses the mechanism for this association, and in particular, whether these susceptibility alleles are involved in the presentation of beryllium to CD4+ T cells. The results clearly indicate that HLA-DP has a selective, although perhaps not exclusive, ability to present beryllium to lung-derived CD4+ T cells from CBD patients compared with HLA-DR and -DQ molecules. Importantly, the DPB1 alleles that mediate presentation in these studies appear to be the same as those involved in disease susceptibility.
The responder CD4+ T cell lines used to determine MHC class II presenting molecules were generally heterogeneous in nature as determined by T cell receptor V region expression, and a multitude of beryllium-reactive clones appeared within these short-term stimulated populations. It is therefore remarkable that the responses of nearly all of these clones to beryllium presented by autologous cells were inhibited by an anti-DP mAb. In CBD patient 1, these blocking data coupled with the studies using allogeneic cells to present beryllium, indicated that essentially all presentation was occurring through HLA-DP. Concomitant studies with two anti-DR reagents also showed inhibition of responses to beryllium, although inhibition was partial and required higher concentrations of the anti-DR reagents. The anti-DR mediated inhibition is difficult to explain. It could not reflect true blocking of DR presentation in CBD patient 1 because this patient was homozygous for DRB1*1101 allele, and this allele (as well as the coinherited DRB3 molecule) did not present beryllium to CD4+ T cell lines from this patient. We wondered whether the inhibition induced by the anti-DR mAbs was somehow an artifact based on crossreactivity with HLA-DP. However, this explanation was shown to be incorrect because presentation by fibroblasts that express only DP was not affected by the anti-DR reagents. Although DRα-DPβ molecules could explain inhibition with the anti-DRα mAbs, studies have shown that such mixed isotype molecules are unlikely to form, let alone be abundantly expressed on the cell surface (19). In one case, although HLA-DP on autologous cells appeared to mediate the majority of beryllium presentation, evidence for presentation by HLA-DR15 was obtained. Of the four patients studied, this was the only DR (or DQ) allele shown to be capable of presenting beryllium. With additional rounds of stimulation, this BAL T cell line was overgrown by dominant Vβ3+ clones with a CDR3 motif previously implicated in disease pathogenesis (11). Interestingly, these CD4+ T cell clones were now totally restricted by HLA-DP, and DR15 was no longer able to present beryllium (data not shown).
A separate role for HLA-DP compared with other class II MHC molecules has yet to be defined (20). Similar to HLA-DR and DQ, this molecule has been shown to be involved in the presentation of peptides to CD4+ T cells (21–24). The present data suggest that particular HLA-DP alleles are distinct in their ability to present beryllium. Interestingly, occupational lung disease related to cobalt exposure also has been associated with inheritance of certain HLA-DP alleles (1), and cobalt has been found to bind purified HLA-DP versus DR molecules (25).
Specific class II HLA-DR, -DQ, and -DP alleles have been associated with susceptibility to immune-mediated diseases, particularly autoimmune diseases (26). For example, the major genetic contribution to rheumatoid arthritis involves DRB1 alleles such as *0401 and *0404 (27), whereas HLA-DQ alleles, especially DQ2 and DQ8, provide the major genetic contribution to type 1 diabetes (26). Certain DP alleles also have been implicated as genetic susceptibility alleles for pauci-articular juvenile chronic arthritis (28), Graves disease (29), and Takayasu's arteritis (30). One central hypothesis for the increased susceptibility to disease is that the associated class II molecule preferentially presents a critical epitope to disease-relevant T cells. However, for essentially all of these disease associations, the mechanism for disease association remains undefined in large part because the initiating autoantigen remains unknown. In the present work, we were able to study an immune-mediated disease with a known HLA-DP allele association and a known pathogenic antigen, which allowed the delineation of the mechanism.
Richeldi et al. (5) reported a negative association between DPB1*0401 and development of CBD after exposure to beryllium, whereas a positive association was noted with DPB1*0201. Additional studies by this group and other investigators showed that DPB1 alleles with a glutamate (E) at position 69, which includes DPB1*0201, were strongly associated with disease susceptibility (5–7). The greatest susceptibility for the development of CBD in exposed individuals occurred in those possessing two Glu-69-containing alleles (7). Based on these studies, HLA-DP typing has been used to identify individuals at risk for CBD. The present studies indicate that these associations with disease susceptibility are most likely based on the ability to present beryllium to pathogenic CD4+ T cells. Overall, we found two alleles, DPB1*0101 and DPB1*0401, that were unable to stimulate the different beryllium-reactive T cell lines after addition of BeSO4 and neither expressed a glutamate (E) at position 69 (Table 2). Among the alleles that did present beryllium, all but one had a glutamate at position 69. One allele, DPB1*0402, was able to present beryllium, but was not previously implicated in disease susceptibility. Our results suggest that this DP allele and the invariant importance of a glutamate at position 69 should be re-evaluated.
A comparison of the amino acid sequences for DPB1 presenting and nonpresenting alleles (Table 2) shows two polymorphic regions with major charge differences. These regions include positions 55–56 and position 69 on the DP β-chain α-helix of the peptide-binding groove, in which the presenting alleles have negatively charged amino acids. Our data suggest that the polymorphism at positions 55–56 may be more important for presentation and possibly susceptibility because every presenting allele showed this difference. Importantly, among frequently inherited HLA-DP alleles, the glutamate at position 69 and the negatively charged residues at positions 55–56 frequently coexist (31), and both may contribute to disease susceptibility. Position 57 in DQβ (corresponding to position 55 in DPβ; ref. 32) also has been associated with susceptibility and resistance to type 1 diabetes (26, 33). This residue can form a salt bridge with a conserved arginine (R) residue at position 79 of the class II α-chain that is critical in the formation of the antigen-binding cleft (33). The shared region among DR4 alleles associated with rheumatoid arthritis includes position 71 (corresponding to position 69 in DPβ) (27).
Beryllium presentation is most likely determined by the ability to bind beryllium, which could occur by direct binding to the MHC molecule or through MHC's binding of particular peptides. Fig. 6 shows a structure of the DPB1*0201 modeled from the DR1 structure. It is important to note that the DPβ polymorphic residues described above correspond to amino acid positions 57–58 and 71 of the DRβ and DQβ chains (32). The polymorphic-charged residues important in beryllium presentation are predicted to be located on the α-helix with side groups pointing into the peptide binding cleft (32, 34), suggesting that they may affect beryllium binding through the peptides bound in the groove and not through a direct interaction with beryllium.
Figure 6.
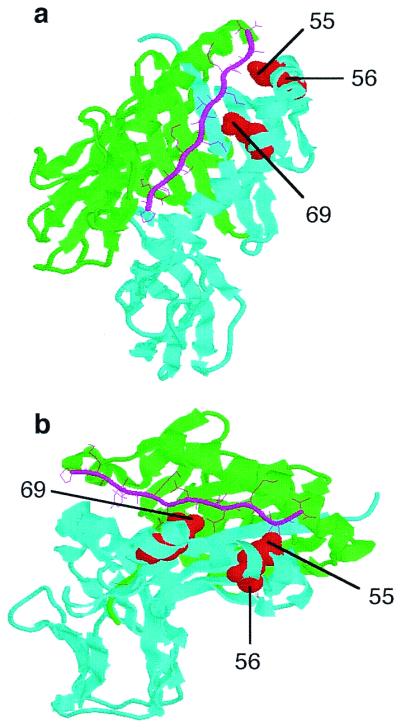
Modeling of the HLA-DP molecule based on the DR1 molecule. (a) Top view of the DR1 peptide-binding cleft, with a green α-chain and a cyan β-chain. (b) Side view of the DR1 peptide binding groove. The HA306–318 peptide is indicated by pink sticks. The polymorphic charged residues at positions 55–56 and 69, indicated as red balls, are located in the peptide binding region. Despite the appearance from the ribbon structure, minimal, if any, portions of these polymorphic residues are exposed on the outside or top of the β-chain.
Acknowledgments
This work is supported by National Institute of Health Grants HL62410, HL06358, and HL03982, General Clinical Research Center Grant M01-RR-0051 from the Division of Research Resources, and Environmental Protection Agency Grant R825702.
Abbreviations
- CBD
chronic beryllium disease
- BAL
bronchoalveolar lavage
- BeSO4
beryllium sulfate
- LCL
lymphoblastoid cell line
- PBMC
peripheral blood mononuclear cell
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.220430797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.220430797
References
- 1.Newman L S, Maier L A, Nemery B. In: Interstitial Lung Disease. Schwarz M I, King T E Jr, editors. Hamilton: Decker; 1998. pp. 367–392. [Google Scholar]
- 2.Kreiss K, Wasserman S, Mroz M M, Newman L S. J Occup Med. 1993;35:267–274. [PubMed] [Google Scholar]
- 3.Kreiss K, Mroz M M, Zhen B, Martyny J W, Newman L S. Am Rev Respir Dis. 1993;148:985–991. doi: 10.1164/ajrccm/148.4_Pt_1.985. [DOI] [PubMed] [Google Scholar]
- 4.Kreiss K, Mroz M M, Newman L S, Martyny J, Zhen B. Am J Ind Med. 1996;30:16–25. doi: 10.1002/(SICI)1097-0274(199607)30:1<16::AID-AJIM3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 5.Richeldi L, Sorrentino R, Saltini C. Science. 1993;262:242–244. doi: 10.1126/science.8105536. [DOI] [PubMed] [Google Scholar]
- 6.Richeldi L, Kreiss K, Mroz M M, Zhen B, Tartoni P, Saltini C. Am J Ind Med. 1997;32:337–340. doi: 10.1002/(sici)1097-0274(199710)32:4<337::aid-ajim3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, White P S, Petrovic M, Tatum O L, Newman L S, Maier L A, Marrone B L. J Immunol. 1999;163:1647–1653. [PubMed] [Google Scholar]
- 8.Newman L S, Bobka C, Schumacher B, Daniloff E, Zhen B, Mroz M, King T E., Jr Am J Respir Crit Care Med. 1994;150:135–142. doi: 10.1164/ajrccm.150.1.8025739. [DOI] [PubMed] [Google Scholar]
- 9.Rossman M D, Kern J A, Elais J A, Cullen M R, Epstein P E, Preuss O P, Markham T N, Daniele R P. Ann Intern Med. 1988;108:687–693. doi: 10.7326/0003-4819-108-5-687. [DOI] [PubMed] [Google Scholar]
- 10.Saltini C, Winestock K, Kirby M, Pinkston P, Crystal R G. N Engl J Med. 1989;320:1103–1109. doi: 10.1056/NEJM198904273201702. [DOI] [PubMed] [Google Scholar]
- 11.Fontenot A P, Falta M T, Freed B M, Newman L S, Kotzin B L. J Immunol. 1999;163:1019–1026. [PubMed] [Google Scholar]
- 12.Saltini C, Amicosante M, Franchi A, Lombardi G, Richeldi L. Eur Respir J. 1998;12:1463–1475. doi: 10.1183/09031936.98.12061463. [DOI] [PubMed] [Google Scholar]
- 13.Newman L S, Kreiss K, King T E, Jr, Seay S, Campbell P A. Am Rev Respir Dis. 1989;139:1479–1486. doi: 10.1164/ajrccm/139.6.1479. [DOI] [PubMed] [Google Scholar]
- 14.Striebich C C, Falta M T, Wang Y, Bill J, Kotzin B L. J Immunol. 1998;161:4428–4436. [PubMed] [Google Scholar]
- 15.Watson A J, DeMars R, Trowbridge I S, Bach F H. Nature (London) 1983;304:358–361. doi: 10.1038/304358a0. [DOI] [PubMed] [Google Scholar]
- 16.Spits H, Borst J, Giphart M, Coligan J, Terhorst C, De Vries J E. Eur J Immunol. 1984;14:299–304. doi: 10.1002/eji.1830140404. [DOI] [PubMed] [Google Scholar]
- 17.Lamarre D, Capon D J, Karp D R, Gregory T, Long E O, Sekaly R P. EMBO J. 1989;8:3271–3277. doi: 10.1002/j.1460-2075.1989.tb08487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosloniec E F, Brand D D, Myers L K, Whittington K B, Gumanovskaya M, Zaller D M, Woods A, Altmann D M, Stuart J M, Kang A H. J Exp Med. 1997;185:1113–1122. doi: 10.1084/jem.185.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karp D R, Teletski C L, Jaraquemada D, Maloy W L, Coligan J E, Long E O. J Exp Med. 1990;171:615–628. doi: 10.1084/jem.171.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer C G, May J, Schnittger L. Immunol Today. 1997;18:58–61. doi: 10.1016/s0167-5699(96)30071-6. [DOI] [PubMed] [Google Scholar]
- 21.Celis E, Ou D, Otvos L., Jr J Immunol. 1988;140:1808–1815. [PubMed] [Google Scholar]
- 22.Celis E, Larson J, Otvos L, Jr, Wunner W H. J Immunol. 1990;145:305–310. [PubMed] [Google Scholar]
- 23.Hammond S A, Obah E, Stanhope P, Monell C R, Strand M, Robbins F M, Bias W B, Karr R W, Koenig S, Siliciano R F. J Immunol. 1991;146:1470–1477. [PubMed] [Google Scholar]
- 24.Kurane I, Dai L C, Livingston P G, Reed E, Ennis F A. J Virol. 1993;67:6285–6288. doi: 10.1128/jvi.67.10.6285-6288.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potolicchio I, Festucci A, Hausler P, Sorrentino R. Eur J Immunol. 1999;29:2140–2147. doi: 10.1002/(SICI)1521-4141(199907)29:07<2140::AID-IMMU2140>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 26.Nepom G T, Erlich H. Annu Rev Immunol. 1991;9:493–525. doi: 10.1146/annurev.iy.09.040191.002425. [DOI] [PubMed] [Google Scholar]
- 27.Nepom G T. Adv Immunol. 1998;68:315–332. doi: 10.1016/s0065-2776(08)60563-5. [DOI] [PubMed] [Google Scholar]
- 28.Begovich A B, Bugawan T L, Nepom B S, Klitz W, Nepom G T, Erlich H A. Proc Natl Acad Sci USA. 1989;86:9489–9493. doi: 10.1073/pnas.86.23.9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong R P, Kimura A, Okubo R, Shinagawa H, Tamai H, Nishimura Y, Sasazuki T. Hum Immunol. 1992;35:165–172. doi: 10.1016/0198-8859(92)90101-r. [DOI] [PubMed] [Google Scholar]
- 30.Dong R P, Kimura A, Numano F, Yajima M, Hashimoto Y, Kishi Y, Nishimura Y, Sasazuki T. Tissue Antigens. 1992;39:106–110. doi: 10.1111/j.1399-0039.1992.tb01918.x. [DOI] [PubMed] [Google Scholar]
- 31.Gilchrist F C, Bunce M, Lympany P A, Welsh K I, du Bois R M. Tissue Antigens. 1998;51:51–61. doi: 10.1111/j.1399-0039.1998.tb02946.x. [DOI] [PubMed] [Google Scholar]
- 32.Stern L J, Brown J H, Jardetzky T S, Gorga J C, Urban R G, Strominger J L, Wiley D C. Nature (London) 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 33.Todd J A, Acha-Orbea H, Bell J I, Chao N, Fronek Z, Jacob C O, McDermott M, Sinha A A, Timmerman L, Steinman L, et al. Science. 1988;240:1003–1009. doi: 10.1126/science.3368786. [DOI] [PubMed] [Google Scholar]
- 34.Chicz R M, Graziano D F, Trucco M, Strominger J L, Gorga J C. J Immunol. 1997;159:4935–4942. [PubMed] [Google Scholar]