

Abstract
Aims
To evaluate whether simvastatin influences (i) the intestinal expression of P-glycoprotein (P-gp) and MRP2, and (ii) the disposition of the β1-selective blocker talinolol, a substrate of these transporter proteins.
Methods
The disposition of talinolol after intravenous (30 mg) and single or repeated oral administration (100 mg daily) was monitored before and after chronic treatment with simvastatin (40 mg daily) in 18 healthy subjects (10 males, eight females, body mass index 19.0–27.0 kg m−2) genotyped for ABCB1, ABCC2 and SLCO1B1 polymorphisms. The steady-state pharmacokinetics of simvastatin was evaluated before and after repeated oral talinolol administration. The duodenal expression of ABCB1 and ABCC2 mRNA before and after simvastatin treatment was quantified using real-time reverse transcriptase-polymerase chain reaction (TaqMan®).
Results
Simvastatin did not influence the expression of duodenal ABCB1 and ABCC2. There was no significant pharmacokinetic interaction between simvastatin and talinolol. Duodenal ABCB1 mRNA content was significantly correlated with the AUC0–∞ (r = 0.627, P = 0.039) and Cmax (r = 0.718, P = 0.013) of oral talinolol. The ABCB1 and ABCC2 gene polymorphisms did not influence simvastatin and talinolol disposition. The half-life of the latter was significantly shorter in the nine carriers with a SLCO1B1*1b allele compared with the seven subjects with the wild-type SLCO1B1*1a/*1a genotype (12.2 ± 1.6 h vs. 14.5 ± 1.4 h, P = 0.01).
Conclusions
Simvastatin does not influence the intestinal expression of P-gp and MRP2 in man. There was no pharmacokinetic interaction between talinolol and simvastatin during their chronic co-administration to healthy subjects.
Keywords: OATP1B1, MRP2, P-glycoprotein, simvastatin, talinolol
Introduction
Talinolol is a β1-selective adrenoreceptor blocking agent used to treat arterial hypertension and coronary heart disease. The drug is slowly and incompletely absorbed after oral administration despite adequate lipid solubility (logD = 1.1 at pH 7.4 and 37°C) and the absence of significant first-pass metabolism [1, 2]. There is ample evidence that the oral absorption of talinolol is influenced by the multidrug transporter P-glycoprotein (P-gp, expressed by the ABCB1 gene) and the multidrug resistance-related protein 2 (MRP2, expressed by the ABCC2 gene). First, talinolol was shown to be a substrate of P-gp and/or MRP2 using Caco2 cell monolayers, rat inverted intestinal sacs and mdr1a/1b knock-out mice [2–4]. Second, the drug is secreted into gut lumen against a steep concentration gradient [5, 6], and the total body clearance is significantly related to intestinal expression of P-gp [7]. Third, pretreatment of healthy subjects with the P-gp (and MRP2) inducers rifampicin, carbamazepine, thyroxine and St John’s Wort is associated with lower bioavailability of talinolol, whereas co-medication with the P-gp inhibitors erythromycin and D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS, a surfactant in pharmaceutical dosage forms) leads to higher bioavailability [7–12]. Fourth, talinolol is better absorbed and more widely distributed in MPR2-deficient rats (Groningen Yellows, GY/TR–) [13]. However, the intestinal clearances of talinolol after intravenous and oral administration in man have not yet been determined.
During cardiovascular therapy, talinolol may interact with other substrates of P-gp and/or MPR2 as shown for the cardiac glycoside digoxin [14]. Another candidate for interaction with talinolol is the cholesterol-lowering simvastatin, which undergoes intensive presystemic ‘first-pass’ elimination leading to very low bioavailability (<5%). The drug is completely metabolized to β-hydroxy simvastatin acid and four other active metabolites, mainly by CYP3A4, and also in part by CYP2C9 and CYP2D6 [15]. Furthermore, simvastatin seems to be substrate of P-gp, MPR2 and the organic anion uptake transporting protein 1B1 (OATP1B1, expressed by the SLCO1B1 gene) and may compete with the active transport of other drugs [16–20]. Elevation of simvastatin plasma concentrations after treatment with itraconazole, erythromycin or ciclosporin, as well as the decrease in bioavailability after enzyme induction by rifampicin, probably result from an interaction with CYP3A4 and P-gp [21, 22]. Therefore, talinolol might interact with the P-gp-dependent transport of simvastatin. On the other hand, there is some experimental evidence that simvastatin may increase expression of multidrug transporter proteins [23]. The structurally very similar lovastatin was shown to be a strong inducer of PXR, which regulates the expression of CYP3A4, in vitro[24]. It is not known whether simvastatin causes the induction of intestinal efflux transporters in man, which in turn could impair the oral absorption of certain drugs.
In this paper, we describe the basic pharmacokinetics of talinolol including its intestinal clearance and correlations to ABCB1, ABCC2 and SLCO1B1 expression and/or genotype. Furthermore, we confirm the absence of relevant clinical pharmacokinetic drug interactions between talinolol and simvastatin.
Materials and methods
Clinical study protocol
Subjects Eighteen healthy white subjects (10 males, eight females; age 21–30 years; body mass index 19.0–27.0 kg m−2) were selected according to their ABCB1 haplotype [25–27] as follows: 2677GG/3435CC (*1/*1, N = 4), 2677GG/3435CT (*1/*6, N = 3), 2677GT and 3435CT (*1/*3, N = 3), 2677GT/3435TT (*6/*6, N = 1), 2677TT/3435CT (*7/*3, N = 1), 2677TT/3435TT (*3/*3, N = 6). The frequency of the carriers with the *3 allele were higher than in an unselected population. Additional screening for ABCC2 polymorphisms identified six subjects with the CC-allele, 11 subjects with the CT and one subject with the TT allele with respect to the C-24T polymorphism [26, 28]. The following haplotypes of the SLCO1B1 gene [28–30]: *1a/*1a (N = 7), *1a/*1b (N = 5), *1b/*5 (N = 1), *1b/*15 (N = 3) and *1a/*5 (N = 1) were identified.
The subjects were in good health based on medical history, physical examination, 12-lead ECG and bicycle ergometry, and routine clinical chemistry and haematology. All subjects tested negative for drugs, hepatitis virus B and C (HBV, HCV) and human immunodeficiency virus (HIV), took no medication except hormonal contraceptives (females), did not drink alcohol or drank < 25 g per day. Two subjects were smokers (less than 10 cigarettes per day). A standardized diet was served during hospitalization. The study protocol was approved by the local ethics committee (University of Greitswald) and all subjects gave written informed consent.
Study protocol The study with simvastatin and talinolol was performed according to the ICH-GCP guidance following the design shown in Figure 1. After inclusion, 11 subjects who agreed to two duodenal biopsies underwent a gastroduodenoscopy and tissue specimens were taken from the lower duodenal mucosa and immediately transferred into liquid nitrogen for mRNA analysis.
Figure 1.
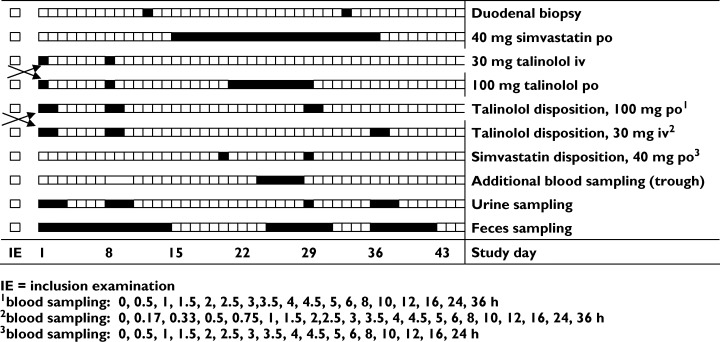
Design of the simvastatin–talinolol interaction study
On the 1st and 8th study day, the pharmacokinetics of talinolol (Cordanum®; AWD.pharma, Germany) was evaluated in the entire group of 18 subjects after intravenous infusion of 30 mg in 200 ml saline (infusion time 30 min) and after oral administration of 100 mg according to a cross-over design with random allocation. Blood was sampled before and 0.17, 0.33, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 10, 12, 16, 24 and 36 h after intravenous infusion and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 10, 12, 16, 24 and 36 h after oral administration. Urine was sampled for 3 days and faeces for 7 days.
Between the 15th and 36th study days, the subjects were treated orally with 40 mg simvastatin once daily (Simvacard®; AWD.pharma), and talinolol (100 mg daily) was coadministered between the 21st and 29th study days. Serum concentration–time profiles of simvastatin were monitored on the 20th study day and those of simvastatin and talinolol on the 29th study day. Venous blood was collected before and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 8, 10, 12, 16, and 24 h (36 h, for the assessment of talinolol half-life) after drug administration. Urine was collected on the 29th study day and faeces between the 25th and 30th study days. Additional trough concentrations were measured as indicated in Figure 1. A second gastroduodenoscopy with duodenal biopsy was performed in the subgroup of 11 subjects at the end of the treatment period with simvastatin (34/35th study day).
Serum and aliquots of urine and faeces were stored at − 80 °C until the analysis. On blood sampling days, standard meals were given 5, 8 and 11 h after administration of the study medication. A standard breakfast during chronic treatment with talinolol and simvastatin was given 1 h after the respective morning administration of the drugs. The meals did not include food constituents that are known to inhibit or induce the function of the transporter proteins investigated.
Drug and metabolite analysis
Simvastatin and β-hydroxy simvastatin acid were assayed using a modified LC-MS/MS method [31, 32]. Simvastatin was obtained from Medinsa (Madrid, Spain) and simvastatin acid from Synfine Research Inc. (Richmond Hill, Canada). Sample processing was carried out in an ice-water bath and as rapidly as possible. In brief, 0.5 ml serum and 25 µl of the internal standard solutions (160 ng ml−1 lovastatin and lovastatin acid) were added to 0.2 ml ammonium acetate buffer (100 mm, pH 5.0). Lovastatin was purchased from Sigma-Aldrich (Steinheim, Germany) and lovastatin acid form Synfine Research Inc. The analytes were extracted with 4 ml methyl tert-butyl ether. The organic layer was evaporated to dryness at room temperature and the residue was dissolved in 100 µl of mobile phase (1 mm ammonium acetate buffer, pH 4.5) of which 70 µl was injected onto the precolumn XTerra® C18 MS, 2.1–10 mm, 3.5 µm and the analytical column XTerra® C18 MS, 2.1–100, 3.5 µm (Waters, Eschborn, Germany). The autosampler was precooled to 4°C using the Peltier cooling system (Perkin Elmer, Rodgau-Jügesheim, Deutschland). For detection, the PE Sciex API 2000 tandem mass spectrometer with TurboIon® spray (Applied Biosystems, Darmstadt, Germany) was used in positive MRM mode for simvastatin (m/z 419 → m/z 199) and lovastatin (m/z 405 → m/z 199) and in negative mode for simvastatin acid (m/z 435 → m/z 319) and lovastatin acid (m/z 421 → m/z 319). The limit of quantification for simvastatin and simvastatin acid was 0.05 ng ml−1. Between day accuracy was − 5.8 to 3.5% (simvastatin) and 1.9–6.2% (simvastatin acid) of the nominal values. The results for between-day precision were 7.5–9.2% and 7.1–8.0%, respectively.
Talinolol in serum, urine and faeces was assayed by a high-performance liquid chromatography method with fluorescence detection [14, 33]. The limits of quantification for talinolol in serum and urine were 5 ng ml−1 and 25 ng ml−1 in faeces. For talinolol in serum, the accuracy of the assay was − 0.3 to 4.6% of the nominal values, and the within-day and between-day precision was 1.1–12.0% and 5.5–7.0%, respectively. For the talinolol in urine, the accuracy was − 4.2 to 0.3% of the nominal values, and the within-day and between-day precision was 3.6–10.0% and 7.3–9.7%, respectively. The respective values for faeces were 2.6–6.1% (accuracy), 3.3–14.6% and 7.0–13.8% (within-day and between day precision).
The excretion of talinolol and its metabolites 2-cis, 3-cis, 3-trans, 4-cis and 4-trans hydroxy talinolol into urine and faeces was quantified using LC-MS/MS, and with a XTerra® C18 MS, 2.1–10 mm, 3.5 µm pre-column and a XTerra® C18 MS, 2.1–100, 3.5 µm analytical column (Waters, Eschborn, Germany) was used. Detection was with a PE Sciex API 2000 mass spectrometer (Applied Biosystems). The TurboIon® spray (400 °C) was used in positive MRM mode at mass transitions of m/z 380 → m/z 324 for all metabolites and m/z 300 → m/z 227 m/z for the internal standard metoclopramide. The limit of quantification for all talinolol metabolites in urine and faeces was 5 ng ml−1. Accuracy was between − 11.5 and 5.6%. Within-day and between-day precision was 0.1–20.2% and 2.3–15.7%, respectively.
Pharmacokinetic and statistical analysis
Maximum plasma concentrations (Cmax, Css,max), minimum concentrations (Cmin, Css,min) and the time to Cmax (tmax) were taken from the concentration–time curves. Average steady-state concentration was obtained from the expression Cav = AUC0–τ /τ (τ = dosage interval). The area under the serum concentration–time curve during an administration interval (AUC0–τ) was calculated using the trapezoidal rule with nontransformed concentration values. AUC0–∞ was determined up to the last sampling time above the limit of quantification (AUC0–t) using the trapezoidal formula and extrapolated to infinity using the concentrations of the terminal slope after logarithmic transformation (AUCt–∞). The elimination half-life (t1/2) was also estimated from logarithmic values of the terminal data points. Total body clearance (CL) was obtained by dividng the intravenous dose by the AUC0–∞. Bioavailability (F) was determined from the expression AUC0–τ × CL/doseoral. Renal clearance (CLR), metabolic clearance (CLM) and intestinal clearance (CLintestinal) were derived from the amounts (Ae) excreted into the urine and faeces divided by the respective AUC0–∞ (single dose) and AUC0–τ (steady state), respectively. Peak–trough fluctuation (PTF) were obtained from the equation PTF = Css,max − Css,min/Cav, and peak–trough swing (PTS) from PTS = Css,max − Css,min/Cmin.
The absence of a clinically relevant influence of talinolol on simvastatin disposition was assumed if the 90% confidence interval on the AUC0−24 h and PTF ratios for simvastatin given before compared with after talinolol was within the range 0.80–1.25. Similarly, the absence of an influence of simvastatin on talinolol disposition was assumed if the AUC0−24 h (steady state after co-medication of simvastatin) to AUC0–∞ (single dose administration before simvastatin) ratio was within the range 0.80–1.25.
Geometric means and geometric standard deviations are present for all concentration and pharmacokinetic data, except F and tmax for which arithmetic means and standard deviations (M ± SD) were determined. Evaluation of the effect of ABCB1, ABCC2 and OATP1B1 genotypes was based on box plots with median and quartiles. Correlations were performed using the Spearman rank test. Gene–dose effects were evaluated using the Kruskal–Wallis test and differences were assessed using the Wilcoxon and Mann–Whitney tests, as appropriate.
Results
After an intravenous infusion of 30 mg of talinolol maximum serum concentrations between 340 and 780 ng ml−1 were achieved. The drug was eliminated with a terminal half-life of about 13 h (Figure 2). The mean renal clearance was somewhat higher than the normal creatinine clearance of healthy young subjects. The metabolic clearance was less than 3% and the intestinal clearance about 20% of the total body clearance. Nearly 50% of the dose was excreted with the urine, and about 25% was recovered unchanged in the faeces (Table 1).
Figure 2.
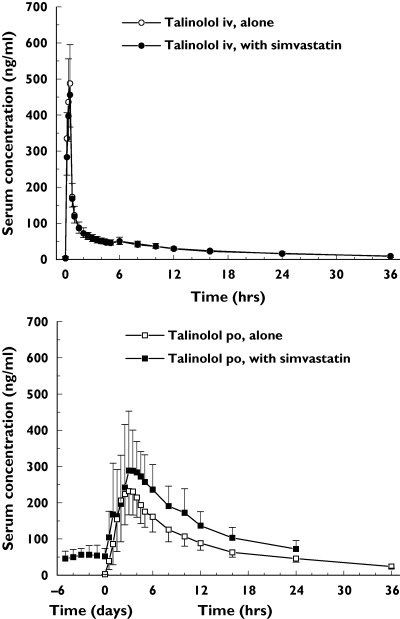
The concentration–time profiles (geometric means and standard deviations) of talinolol after infusion of 30 mg before and during repeated co-medication of 40 mg simvastatin daily (top panel). The concentration–time curves of talinolol after single oral administration alone and after chronic oral administration of 100 mg daily together with 40 mg simvastatin daily are given. The study was performed in 18 healthy subjects (bottom panel)
Table 1.
Pharmacokinetic parameters of talinolol after intravenous and chronic oral administration and during co-medication with simvastatin
| Intravenous talinolol (30 mg) | Oral talinolol (100 mg) | Point estimates (90% CI) | |||||
|---|---|---|---|---|---|---|---|
| Without simvastatin | With simvastatin | Single dose without simvastatin | Steady-state with simvastatin | Intravenous talinolol | Oral talinolol | ||
| AUC0–∞* | (ng h−1 ml−1) | 1406 (1167, 1695) |
1405 (1210, 1632) |
3438 (2667, 4433) |
3674 (2835, 4761) |
0.999 (0.946, 1.056) |
1.069 (0.997, 1.145) |
| AUCt-∞ | (%) | 12.1 ± 2.8 | 12.7 ± 3.6 | 13.3 ± 3.4 | – | – | – |
| Cmax* | (ng/ml) | 496 (401, 613) |
464 (377, 570) |
337 (235, 483) |
382 (281, 520) |
0.936 (0.879, 0.997) |
– – |
| tmax | (h) | 0.46 ± 0.09 | 0.48 ± 0.05 | 2.69 ± 0.97 | 3.53 ± 2.02 | 0.019 (− 0.014, 0.051) |
0.833 (− 0.008, 1.674) |
| F | (%) | – | – | 73.3 (62.5, 86.0) |
78.4 (64.1, 95.9) |
1.069 (0.983, 1.164) |
|
| t1/2 | (h) | 13.5 (12.1, 15.1) |
13.3 (11.3, 15.7) |
13.0 (11.3, 15.0) |
13.9 (11.8, 16.5) |
0.988 (0.929, 1.051) |
1.073 (0.997, 1.153) |
| CL | (ml min−1) | 356 (295, 429) |
356 (306, 413) |
– | – | 1.001 (0.947, 1.057) |
– – |
| CLR | (ml min−1) | 152 (94, 246) |
167 (94, 297) |
148 (110, 199) |
153 (106, 221) |
1.102 (0.803, 1.511) |
1.029 (0.885, 1.197) |
| CLM | (ml min−1) | 7.26 (3.95, 13.37) |
8.02 (5.45, 11.81) |
9.57 (7.16, 12.78) |
8.11 (5.40, 12.19) |
1.105 (0.796, 1.533) |
0.848 (0.730, 0.986) |
| CLintestinal | (ml min−1) | 74 (37, 147) |
100 (62, 162) |
146 (68, 315) |
175 (94, 324) |
1.356 (1.089, 1.689) |
1.197 (0.884, 1.620) |
| Aeurine | (mg) | 13.0 (8.9, 19.1) |
14.4 (9.0, 23.2) |
31.2 (23.8, 41.0) |
34.5 (23.6, 50.4) |
1.109 (0.838, 1.467) |
1.106 (0.921, 1.327) |
| Aefaces | (mg) | 6.3 (3.5, 11.3) |
8.5 (5.6, 13.0) |
31.6 (17.2, 58.1) |
39.1 (24.4, 62.5) |
1.345 (1.120, 1.617) |
1.235 (0.937, 1.630) |
Geometric means and geometric standard deviations are given for all parameters except tmax, for which arithmetic means and standard deviations are listed. Point estimates and 90% confidence intervals were assessed for the evaluation of equivalence after co-medication with simvastatin.
AUC0−24 h. †Css, max in case of repeated oral administration of talinolol.
After a single oral dose, talinolol was rapidly but incompletely absorbed with a bioavailability of about 75%. Metabolic and renal clearance after oral administration were similar to the respective values after intravenous administration. About 50% of the excreted metabolites were detected in urine, the remaining being present in the faeces. The main metabolite was 4-trans hydroxy talinolol (Table 2). The apparent intestinal clearance of talinolol after oral administration was about twice that after intravenous infusion, suggesting significant presystemic elimination. On average, about 30% of the oral dose was excreted with the urine, nearly 40% in the faeces.
Table 2.
Metabolic clearances of talinolol and the amounts of its hydroxy metabolites excreted into urine and faeces before and during simvastatin co-medication are given
| Intravenous talinolol (30 mg) | Oral talinolol (100 mg) | ||||
|---|---|---|---|---|---|
| Without simvastatin | With simvastatin | Single dose without simvastatin | Steady state with simvastatin | ||
| CLM(faeces) | (ml min−1) | 4.97 (2.52, 9.77) |
5.28 (3.30, 8.45) |
6.70 (4.95, 9.07) |
4.64* (2.61, 8.27) |
| CLM(urine) | (ml min−1) | 1.95 (1.13, 3.38) |
2.48 (1.72, 3.58) |
2.83 (1.90, 4.21) |
3.18 (2.10, 4.82) |
| Amounts excreted into urine and faeces | |||||
| 2-cis-OH | (mg) | 0.033 (0.021, 0.052) |
0.037 (0.022, 0.061) |
0.107 (0.072, 0.160) |
0.078 (0.044, 0.140) |
| 3-cis-OH | (mg) | 0.175 (0.091, 0.336) |
0.176 (0.109, 0.285) |
0.494 (0.314, 0.777) |
0.440 (0.299, 0.648) |
| 3-trans-OH | (mg) | 0.057 (0.033, 0.098) |
0.070 (0.045, 0.108) |
0.168 (0.112, 0.252) |
0.169 (0.117, 0.243) |
| 4-cis-OH | (mg) | 0.040 (0.026, 0.061) |
0.051 (0.027, 0.096) |
0.204 (0.127, 0.330) |
0.133* (0.082, 0.216) |
| 4-trans-OH | (mg) | 0.289 (0.172, 0.487) |
0.312 (0.208, 0.466) |
0.981 (0.702, 1.371) |
0.945 (0.686, 1.301) |
Data are geometric means (geometric standard deviations).
*P < 0.05 compared with data after single-dose administrations of talinolol without simvastatin (Wilcoxon test).
Chronic co-medication of simvastatin did not significantly influence the steady-state pharmacokinetics of talinolol. The 90% confidence intervals on the point estimates of all the pharmacokinetic parameters were within the range of equivalence of 0.80–1.25 (Table 1). Only the amount of 4-cis hydroxy talinolol excreted into the faeces, and hence the metabolic clearance into faeces was significantly decreased after simvastatin co-medication (Table 2).
The mean steady-state serum concentration–time profiles of simvastatin before and during co-medication with talinolol were almost identical (Figure 3), and the corresponding AUC0−24h values were equivalent (point estimate 1.030; 90% CI 0.882, 1.203). However, the drug was more slowly absorbed during talinolol administration based on a significantly decreased PTF. There was also a tendency for a lower Cmax and longer tmax, but the 90% confidence intervals of the respective ratios were not within the stipulated limit of equivalence (Table 3). Talinolol co-medication also did not markedly influence the concentration–time profiles of simvastatin acid, the 90% confidence intervals of the ratios of its pharmacokinetic parameters except PTS being within the range of 0.80–1.25 (Figure 3, Table 3).
Figure 3.
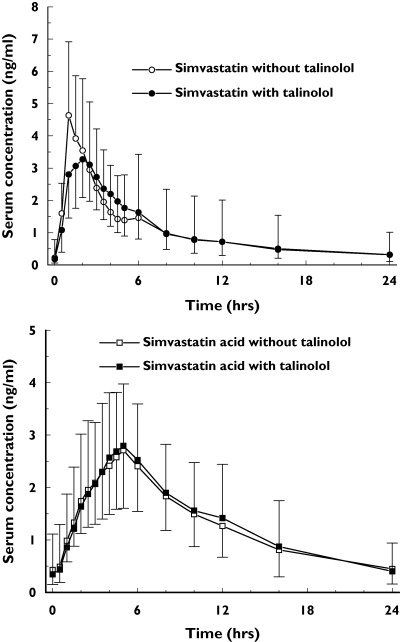
The concentration–time profiles (geometric means and standard deviations) of simvastatin (top panel) and simvastatin acid (bottom panel) after chronic treatment with 40 mg simvastatin daily before and during co-medication of 100 mg talinolol daily in 18 healthy subjects
Table 3.
Pharmacokinetic parameters (geometric mean and standard deviation) of simvastatin and hydroxy simvastatin acid after chronic treatment with 40 mg daily without and during co-medication of 100 mg talinolol.
| Simvastatin | Simvastatin acid | Point estimates (90% CI) | |||||
|---|---|---|---|---|---|---|---|
| Without talinolol | With talinolol | Without talinolol | With talinolol | Simvastatin | Simvastatin acid | ||
| AUC0–24 | (ng h−1 ml−1) | 27.8 (14.8, 52.3) |
28.2 (16.9, 47.1) |
32.4 (18.8, 56.1) |
33.4 (22.4, 49.9) |
1.015 (0.866, 1.191) |
1.030 (0.882, 1.203) |
| PTF | (%) | 4.46 (2.54, 7.83) |
3.68* (2.29, 5.91) |
2.96 (1.81, 4.86) |
2.96 (2.07, 4.24) |
0.825 (0.699, 0.973) |
1.000 (0.847, 1.180) |
| Css,max | (ng ml−1) | 5.47 (3.85, 7.76) |
4.60 (3.08, 6.88) |
0.35 (0.17, 0.71) |
0.29 (0.12, 0.67) |
0.842 (0.723, 0.980) |
0.823 (0.621, 1.090) |
| Css,min | (ng ml−1) | 0.23 (0.10, 0.51) |
0.18 (0.08, 0.42) |
1.35 (0.78, 2.34) |
1.39 (0.93, 2.08) |
0.800 (0.585, 1.095) |
1.030 (0.882, 1.203) |
| Cav | (ng ml−1) | 1.16 (0.62, 2.18) |
1.18 (0.71, 1.96) |
1.87 (1.28, 2.73) |
1.84 (1.35, 2.52) |
1.015 (0.866, 1.191) |
0.986 (0.863, 1.128) |
| PTS | (%) | 22.8 (9.90, 52.6) |
23.9 (9.02, 63.3) |
7.18 (3.34, 15.44) |
8.86 (3.61, 21.77) |
1.047 (0.757, 1.448) |
1.2349 (0.919, 1.661) |
Point estimates and 90% confidence intervals are presented for the comparison of simvastatin disposition when given alone and in the presence of talinolol.
*P < 0.05 compared with data after administration of simvastatin without talinolol co-medication (Wilcoxon test).
A power assessment of the equivalence decision with a sample size of N = 18, an α error of 5%, an equivalence interval of 0.80–1.25 and the true ratios of the point estimates (R = µTest/µReference) and the true intrasubject coefficients (IC) from the study, revealed the following values (nQuery Adviser 5.0, StatSol, Cork, Ireland): 84% for AUC of simvastatin (R = 1.03, IC = 28%), 84% for the PTF of simvastatin (R = 1.00, IC = 29%) and 99% for the AUC of talinolol (R = 1.069, IC = 12%).
The intestinal expression of ABCB1 mRNA (relative to 18S mRNA × 10−3, geometric means and geometric standard deviations) before and after co-medication of simvastatin was 6.04 (4.83, 15.5) vs. 5.02 (3.50. 9.52; not significant). The respective values for ABCC2 mRNA expression were 3.27 (2.06, 5.99) and 2.98 (1.50, 3.83, not significant). There were significant correlations between intestinal ABCB1 mRNA content and the AUC0–∞ as well as the Cmax of talinolol after oral administration (Figure 4). Polymorphisms of the ABCB1 and ABCC2 were considered not to be of functional relevance for the expression of intestinal P-gp and MRP2 or for the disposition of talinolol and simvastatin (data not shown). Talinolol half-lives were significantly shorter in the nine carriers of the SLCO1B1*1b allele compared with the seven subjects with the wild-type SLCO1B1*1a/*1a genotype (12.2 ± 1.6 h vs. 14.5 ± 1.4 h, P = 0.01, Figure 5). There was a trend for a higher faecal excretion of talinolol in subjects with the SLCO1B1*1b allele.
Figure 4.
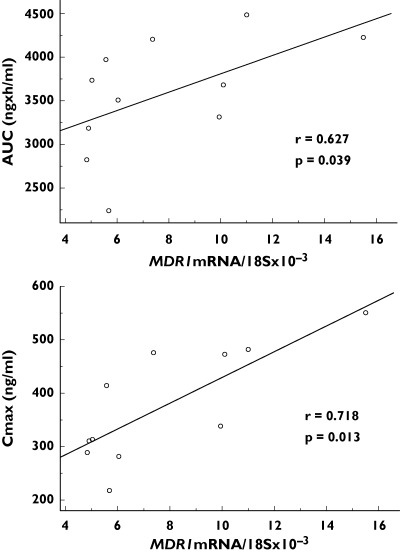
Correlations between intestinal expression of ABCB1 mRNA and the AUC0–∞ and Cmax of talinolol after chronic oral administration in 11 healthy subjects
Figure 5.
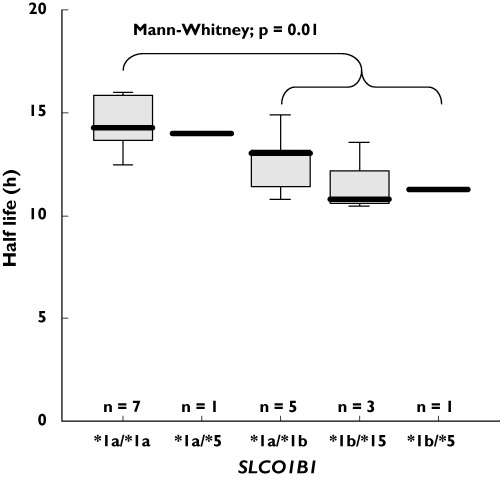
Box plots of the elimination half-lives of talinolol after single oral administration of 100 mg in relation to SLCO1B1 genotype
Discussion
This paper presents the first data on both the renal and faecal elimination of the β1-selective blocker talinolol, which is a probe drug to measure intestinal efflux by P-gp and/or MRP2 [7–12, 14]. The drug was found to be slowly and incompletely absorbed, widely distributed and bi-exponentially eliminated with a terminal half-life of approximately 12–13 h. The renal clearance was greater than creatinine clearance in the young healthy subjects studied, indicating tubular secretion. The metabolic clearance of talinolol, for which CYP3A4 is mainly responsible, was less than 3% of the total body clearance, and the metabolites were excreted in equal amounts into urine and faeces [33]. The intestinal clearance of talinolol after intravenous administration was about 20% of the total body clearance and 50% of the renal clearance. Apparent intestinal clearance after oral administration was about twice as high as that after intravenous administration. Intestinal clearance results from the net elimination of talinolol via the faeces, which may result from biliary and/or intestinal excretion counteracted by intestinal metabolism and/or intestinal re-absorption. The apparent intestinal clearance after oral administration of talinolol may also include a contribution form a non-absorbed portion of the administered dose. However, in our view the faecal excretion of talinolol after intravenous administration and the limited bioavailability of the drug after oral administration may be explained by intestinal secretion via the efflux transporter proteins P-gp and MRP2, of which talinolol is a substrate [2, 4–7]. Unexpectedly, we found a positive correlation between intestinal ABCB1mRNA expression and AUC0–∞ of oral talinolol in our non-induced healthy subjects. Furthermore, talinolol absorption was not dependent on polymorphisms of the ABCB1 and/or ABCC2 genes.
These findings show the complexity of talinolol pharmacokinetics [34]. On the one hand, upregulation of intestinal efflux by rifampicin, carbamazepine, thyroxine and St John’s Wort results in lower oral absorption of talinolol [7–10]. On the other hand, co-medication of verapamil, an inhibitor of P-gp and MRP2, also leads to lower serum concentrations of oral talinolol in mdr1a/1b knock-out and wild-type mice and in healthy subjects. In the latter case an increase in concentration would be expected, at least in species with intact intestinal efflux transporters [3, 35]. Very recently, significantly decreased talinolol bioavailability without change in intestinal P-gp expression was observed after grapefruit juice ingestion [36]. The authors of this work explained their findings on the presence of an unknown organic anion uptake transporter protein (OATP), which is inhibited by verapamil and grapefruit constituents and which overshadows the efflux of talinolol mediated by P-gp and/or MRP2. One might speculate that these OATPs in non-induced subjects are coregulated with intestinal P-gp and/or MRP2. However, it is not known how talinolol is taken up from gut lumen and how this mechanism interacts with the intestinal efflux of the drug.
We found some evidence that the liver-specific uptake transporter OATP1B1 might be involved in the disposition of talinolol. In carriers of the SLCO1B1*1b allele, the half-life of the drug was significantly shorter and faecal excretion tended to be increased. Talinolol in carriers with the *1b variant probably undergoes better hepatic uptake, leading to more efficient secretion into bile by canalicular P-gp and/or MRP2. The protein expressed by SLCO1B1*1b has a higher activity than that by the wild-type gene, as shown by oestrone sulphate uptake in SLCO1B1*1b-transfected HeLa cells and by the results of a pharmacokinetic study with pravastatin in healthy subjects with the *1b allele [30, 37]. However, the present results are difficult to interpret because talinolol undergoes little biliary excretion [38]. Therefore, in vitro data on the binding of the drug to OATP1B1, and pharmacokinetic data from a larger number of subjects are needed to confirm the role of the SLCO1B1 gene polymorphism in the disposition of talinolol.
In treatment of cardiovascular diseases, talinolol is frequently given in combination with other drugs that are subject to CYP3A4 metabolism and/or active membrane transport. One example is the cholesterol-lowering statin simvastatin. This drug may compete with talinolol for intestinal and hepatic CYP3A4, P-gp and MRP2, and probably with hepatic OATP1B1, for which simvastatin is a substrate [16–20]. Since simvastatin undergoes substantial presystemic elimination, markedly increased blood concentrations are expected during co-administration with talinolol, also a substrate of P-gp, MRP2 and CYP3A4. The observed changes in talinolol disposition were assumed to be much lower because of its comparably low presystemic elimination. Furthermore, we hypothesized that simvastatin may affect the disposition of talinolol by induction of intestinal efflux transporters. Simvastatin is similar in structure to the PXR-inducer lovastatin and it regulates expression of mdr2 and mdr1b in rats [23].
In conclusion, the pharmacokinetics of talinolol and simvastatin were not affected during co-administration of these drugs to healthy subjects.
The work was supported by the German Federal Ministry for Education and Research (grant 01ZZ0403) and a research grant of AWD.pharma GmbH & Co. KG Dresden.
References
- 1.Trausch B, Oertel R, Richter K, Gramatte T. Disposition and bioavailability of the beta 1-adrenoceptor antagonist talinolol in man. Biopharm Drug Dispos. 1995;16:403–14. doi: 10.1002/bdd.2510160505. [DOI] [PubMed] [Google Scholar]
- 2.Wetterich U, Spahn-Langguth H, Mutschler E, Terhaag B, Rosch W, Langguth P. Evidence for intestinal secretion as an additional clearance pathway of talinolol enantiomers. concentration-and dose-dependent absorption in vitro and in vivo. Pharm Res. 1996;13:514–22. doi: 10.1023/a:1016029601311. [DOI] [PubMed] [Google Scholar]
- 3.Schwarz UI, Dresser GK, Oertel R. Talinolol–verapamil interaction is not solely due to p-glycoprotein inhibition. Clin Pharmacol Ther. 2001;69:PIII–86. [Google Scholar]
- 4.Spahn-Langguth H, Baktir G, Radschuweit A, Okyar A, Terhaag B, Ader P, Hanafy A, Langguth P. P-glycoprotein transporters and the gastrointestinal tract: evaluation of the potential in vivo relevance of in vitro data employing talinolol as model compound. Int J Clin Pharmacol Ther. 1998;36:16–24. [PubMed] [Google Scholar]
- 5.Gramatte T, Oertel R, Terhaag B, Kirch W. Direct demonstration of small intestinal secretion and site-dependent absorption of the beta-blocker talinolol in humans. Clin Pharmacol Ther. 1996;59:541–9. doi: 10.1016/S0009-9236(96)90182-4. [DOI] [PubMed] [Google Scholar]
- 6.Gramatte T, Oertel R. Intestinal secretion of intravenous talinolol is inhibited by luminal R-verapamil. Clin Pharmacol Ther. 1999;66:239–45. doi: 10.1016/S0009-9236(99)70031-7. [DOI] [PubMed] [Google Scholar]
- 7.Westphal K, Weinbrenner A, Zschiesche M, Franke G, Knoke M, Oertel R, Fritz P, von Richter O, Warzok R, Hachenberg T, Kauffmann HM, Schrenk D, Terhaag B, Kroemer HK, Siegmund W. Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: a new type of drug/drug interaction. Clin Pharmacol Ther. 2000;68:345–55. doi: 10.1067/mcp.2000.109797. [DOI] [PubMed] [Google Scholar]
- 8.Giessmann T, May K, Modess C, Wegner D, Hecker U, Zschiesche M, Dazert P, Grube M, Schroeder E, Warzok R, Cascorbi I, Kroemer HK, Siegmund W. Carbamazepine regulates intestinal P-glycoprotein and multidrug resistance protein MRP2 and influences disposition of talinolol in humans. Clin Pharmacol Ther. 2004;76:192–200. doi: 10.1016/j.clpt.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 9.Siegmund W, Altmannsberger S, Paneitz A, Hecker U, Zschiesche M, Franke G, Meng W, Warzok R, Schroeder E, Sperker B, Terhaag B, Cascorbi I, Kroemer HK. Effect of levothyroxine administration on intestinal P-glycoprotein expression: consequences for drug disposition. Clin Pharmacol Ther. 2002;72:256–64. doi: 10.1067/mcp.2002.126706. [DOI] [PubMed] [Google Scholar]
- 10.Schwarz UI, Hanso H, Dresser GK, Kim RB, Fromm MF, Oertel R, Miehlke S, Kirch W, Effect of St. John’s wort on MDR1 expression in small bowel – consequence for oral bioavailability of talinolol. Eur J Clin Pharmacol. 2002;58:S99. [Google Scholar]
- 11.Bogman K, Zysset Y, Degen L, Hopfgartner G, Gutmann H, Alsenz J, Drewe J. P-glycoprotein and surfactants: effect on intestinal talinolol absorption. Clin Pharmacol Ther. 2005;77:24–32. doi: 10.1016/j.clpt.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Schwarz UI, Gramatte T, Krappweis J, Oertel R, Kirch W. P-glycoprotein inhibitor erythromycin increases oral bioavailability of talinolol in humans. Int J Clin Pharmacol Ther. 2000;38:161–7. doi: 10.5414/cpp38161. [DOI] [PubMed] [Google Scholar]
- 13.Bernsdorf A, May K, Kunert-Keil C, Moritz KU, Kroemer HK, Siegmund W. Expression of P-glycoprotein and MRP2 and distribution of talinolol in MRP2-deficient rats (GY/TR) Naunyn Schmiedebergs Arch Pharmacol. 2003;367:R110. [Google Scholar]
- 14.Westphal K, Weinbrenner A, Giessmann T, Stuhr M, Franke G, Zschiesche M, Oertel R, Terhaag B, Kroemer HK, Siegmund W. Oral bioavailability of digoxin is enhanced by talinolol: evidence for involvement of intestinal P-glycoprotein. Clin Pharmacol Ther. 2000;68:6–12. doi: 10.1067/mcp.2000.107579. [DOI] [PubMed] [Google Scholar]
- 15.Mauro VF. Clinical pharmacokinetics and practical applications of simvastatin. Clin Pharmacokinet. 1993;24:195–202. doi: 10.2165/00003088-199324030-00002. [DOI] [PubMed] [Google Scholar]
- 16.Hsiang B, Zhu Y, Wang Z, Wu Y, Sasseville V, Yang WP, Kirchgessner TG. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J Biol Chem. 1999;274:37161–8. doi: 10.1074/jbc.274.52.37161. [DOI] [PubMed] [Google Scholar]
- 17.Bogman K, Peyer AK, Torok M, Kusters E, Drewe J. HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br J Pharmacol. 2001;132:1183–92. doi: 10.1038/sj.bjp.0703920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakaeda T, Takara K, Kakumoto M, Ohmoto N, Nakamura T, Iwaki K, Tanigawara Y, Okumura K. Simvastatin and lovastatin, but not pravastatin, interact with MDR1. J Pharm Pharmacol. 2002;54:419–23. doi: 10.1211/0022357021778493. [DOI] [PubMed] [Google Scholar]
- 19.Chen C, Mireles RJ, Campbell SD, Lin J, Mills JB, Xu JJ, Smolarek TA. Differential interaction of 3-hydroxy-3-methylglutaryl-coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos. 2005;33:537–46. doi: 10.1124/dmd.104.002477. [DOI] [PubMed] [Google Scholar]
- 20.Wang EJ, Casciano CN, Clement RP, Johnson WW. HMG-CoA reductase inhibitors (statins) characterized as direct inhibitors of P-glycoprotein. Pharmaceut Res. 2001;18:800–6. doi: 10.1023/a:1011036428972. [DOI] [PubMed] [Google Scholar]
- 21.Boger RH. Drug interactions of the statins and consequences for drug selection. Int J Clin Pharmacol Ther. 2001;39:369–82. doi: 10.5414/cpp39369. [DOI] [PubMed] [Google Scholar]
- 22.Kyrklund C, Backman JT, Kivisto KT, Neuvonen M, Laitila J, Neuvonen PJ. Rifampin greatly reduces plasma simvastatin and simvastatin acid concentrations. Clin Pharmacol Ther. 2000;68:592–7. doi: 10.1067/mcp.2000.111414. [DOI] [PubMed] [Google Scholar]
- 23.Hooiveld GJEJ, Vos TA, Scheffer GL, Van Goor H, Koning H, Bloks V, Loot AE, Meijer DKF, Jansen PLM, Kuipers F, Muller M. 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) induce hepatic expression of the phospholipid translocase mdr2 in rats. Gastroenterology. 1999;117:678–87. doi: 10.1016/s0016-5085(99)70462-2. [DOI] [PubMed] [Google Scholar]
- 24.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–23. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siegmund W, Ludwig K, Giessmann T, Dazert P, Schroeder E, Sperker B, Warzok R, Kroemer HK, Cascorbi I. The effects of the human MDR1 genotype on the expression of duodenal P-glycoprotein and disposition of the probe drug talinolol. Clin Pharmacol Ther. 2002;72:572–83. doi: 10.1067/mcp.2002.127739. [DOI] [PubMed] [Google Scholar]
- 26.Giessmann T, Modess C, Hecker U, Zschiesche M, Dazert P, Kunert-Keil C, Warzok R, Engel G, Weitschies W, Cascorbi I, Kroemer HK, Siegmund W. CYP2D6 genotype and induction of intestinal drug transporters by rifampin predict presystemic clearance of carvedilol in healthy subjects. Clin Pharmacol Ther. 2004;75:213–22. doi: 10.1016/j.clpt.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 27.Kim RB, Leake BF, Choo EF, Dresser GK, Kubba SV, Schwarz UI, Taylor A, Xie HG, McKinsey J, Zhou S, Lan LB, Schuetz JD, Schuetz EG, Wilkinson GR. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther. 2001;70:189–99. doi: 10.1067/mcp.2001.117412. [DOI] [PubMed] [Google Scholar]
- 28.Niemi M, Schaeffeler E, Lang T, Fromm MF, Neuvonen M, Kyrklund C, Backman JT, Kerb R, Schwab M, Neuvonen PJ, Eichelbaum M, Kivisto KT. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1) Pharmacogenetics. 2004;14:429–40. doi: 10.1097/01.fpc.0000114750.08559.32. [DOI] [PubMed] [Google Scholar]
- 29.Kim RB. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) and genetic variability (single nucleotide polymorphisms) in a hepatic drug uptake transporter: what’s it all about? Clin Pharmacol Ther. 2004;75:381–5. doi: 10.1016/j.clpt.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 30.Mwinyi J, Johne A, Bauer S, Roots I, Gerloff T. Evidence for inverse effects of OATP-C (SLC21A6) 5 and 1b haplotypes on pravastatin kinetics. Clin Pharmacol Ther. 2004;75:415–21. doi: 10.1016/j.clpt.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 31.Backman JT, Kyrklund C, Kivisto KT, Wang JS, Neuvonen PJ. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin Pharmacol Ther. 2000;68:122–9. doi: 10.1067/mcp.2000.108507. [DOI] [PubMed] [Google Scholar]
- 32.Najib NM, Idkaidek N, Adel A, Admour I, Astigarraga REB, De Nucci G, Alam SM, Dham R Qumaruzaman. Pharmacokinetics and bioequivalence evaluation of two simvastatin 40mg tablets (Simvast & Zocor) in healthy human volunteers. Biopharmaceutics Drug Disposition. 2003;24:183–9. doi: 10.1002/bdd.347. [DOI] [PubMed] [Google Scholar]
- 33.Zschiesche M, Lemma GL, Klebingat KJ, Franke G, Terhaag B, Hoffmann A, Gramatte T, Kroemer HK, Siegmund W. Stereoselective disposition of talinolol in man. J Pharm Sci. 2002;91:303–11. doi: 10.1002/jps.10054. [DOI] [PubMed] [Google Scholar]
- 34.Weitschies W, Bernsdorf A, Giessmann T, Zschiesche M, Modess C, Hartmann V, Mrazek C, Wegner D, Nagel S, Siegmund W. The talinolol double-peak phenomenon is likely caused by presystemic processing after uptake from gut lumen. Pharm Res. 2005;22:728–35. doi: 10.1007/s11095-005-2588-5. [DOI] [PubMed] [Google Scholar]
- 35.Schwarz UI, Gramatte T, Krappweis J, Berndt A, Oertel R, von Richter O, Kirch W. Unexpected effect of verapamil on oral bioavailability of the beta-blocker talinolol in humans. Clin Pharmacol Ther. 1999;65:283–90. doi: 10.1016/S0009-9236(99)70107-4. [DOI] [PubMed] [Google Scholar]
- 36.Schwarz UI, Seemann R, Oertel R, Miehlke S, Kuhlisch E, Fromm MF, Kim RB, Bailey DG, Kirch W. Grapefruit juice ingestion reduces talinolol serum concentration. Clin Pharmacol Ther. 2005;77:OI–B-1. doi: 10.1016/j.clpt.2004.11.111. [DOI] [PubMed] [Google Scholar]
- 37.Tirona RG, Leake BF, Merino G, Kim RB. Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European-and African-Americans. J Biol Chem. 2001;276:35669–75. doi: 10.1074/jbc.M103792200. [DOI] [PubMed] [Google Scholar]
- 38.Terhaag B, Gramatte T, Richter K, Voss J, Feller K. The biliary elimination of the selective beta-receptor blocking drug talinolol in man. Int J Clin Pharmacol Ther Toxicol. 1989;27:170–2. [PubMed] [Google Scholar]