

Abstract
Granulocyte colony-stimulating factor (G-CSF) is essential for the host response to bacterial infection by controlling neutrophil production in the bone marrow. The G-CSF receptor (G-CSFR) activates the Jak/STAT pathway, although little is understood about how these signals regulate basal and stress-induced granulopoiesis. We examined STAT3 function in granulocytes using a bone marrow conditional knockout mouse model. Our results show that STAT3 has a crucial role in emergency granulopoiesis and mature neutrophil function. STAT3-deficient mice have an aberrant response to G-CSF in vivo, characterized by failure to accumulate immature granulocytes and an increased ratio of mature to immature neutrophils in the bone marrow, peripheral blood, and spleen. Acute neutrophil mobilization is impaired in STAT3-deficient mice as judged by their failure to up-regulate circulating neutrophils following short-term G-CSF exposure. STAT3 also controls neutrophil chemotactic responses to natural ligands for CXCR2 and regulates the magnitude of chemoattractant-induced actin polymerization. These functions of STAT3 are independent of its principal target gene Socs3, which encodes a crucial feedback inhibitor of cytokine signaling. Our results demonstrate the existence of distinct STAT3 target pathways in neutrophils required for granulopoiesis and innate immunity.
Introduction
The essential role of neutrophils in antibacterial immunity is shown by the severity of congenital, acquired, and therapy-induced neutropenias, which cause extreme susceptibility to life-threatening infections. The hematopoietic cytokine G-CSF and its receptor (G-CSFR) regulate neutrophil production.1-3 In steady-state hematopoiesis, G-CSF controls viability of the reserve bone marrow neutrophil pool.2-4 Circulating G-CSF levels rise dramatically during infection, stimulating acute neutrophil mobilization from the bone marrow and expansion of the immature granulocyte population to increase neutrophil reserves.5 G-CSF functions have been widely exploited in the clinic to treat neutropenia, particularly in cancer patients following ablative chemotherapy. G-CSF is also used as a potent hematopoietic progenitor mobilization agent for bone marrow transplantation.
Mice lacking the G-CSFR form granulocytes at lower amounts in the bone marrow, and neutrophils from these animals are defective in certain mature cell functions.2,6 Gene-targeting studies with a chimeric G-CSFR showed that the erythropoietin receptor (EpoR) cytoplasmic domain failed to substitute functionally for the G-CSFR intracellular region,7 indicating that G-CSFR selectively activates pathways necessary for terminal granulopoiesis and does not merely provide generic cytokine signals. The molecular pathways downstream of the G-CSFR that are important for controlling neutrophil maturation are largely unknown.
G-CSF stimulates 3 members of the STAT family (STATs 1, 3, and 5), which serve as major cytokine-response proteins that directly regulate gene expression.8 Evidence linking either STAT1 or STAT5 to G-CSF function is limited. Deletion of STAT5a and STAT5b suppressed myeloid repopulating activity and granulocytic progenitor levels in bone marrow, suggesting a potential role in the neutrophil lineage, although further work is warranted because the mouse model employed in these studies expresses a truncated STAT5 protein in certain tissues.9-12 STAT1 does not appear to play a major role in granulocytes.13,14 By contrast, STAT3 has important negative regulatory activity in basal granulopoiesis. Mice lacking STAT3 expression in bone marrow progenitors display marked peripheral neutrophilia under resting conditions.15-17 The role of STAT3 in regulating steady-state neutrophil production is linked to its control of SOCS3, a crucial feedback inhibitor of G-CSF signaling.15,18 Bone marrow–specific deletion of SOCS3 causes neutrophilia in aging mice under basal conditions. SOCS3 deficiency also renders an exaggerated emergency granulopoiesis response to G-CSF characterized by elevated neutrophilia and inflammatory neutrophil tissue infiltration.18
The negative regulatory role of STAT3 in basal granulopoieis was consistent with its control of SOCS3 expression; however, this did not entirely reconcile with studies of G-CSFR signaling function in mice or the requirement for STAT3 in G-CSF–dependent differentiation of granulocytic cell lines.19-23 Moreover, the function of STAT3 in the emergency granulopoiesis response is unknown. Experiments described in this paper for the first time reveal the positive effects of STAT3 in the granulocytic lineage. Mice lacking STAT3 in bone marrow (STAT3-deficient) have an altered response to pharmacologic levels of G-CSF, treatments that mimic emergency granulopoiesis, and therapeutic use of G-CSF. The impairments include a failure to elicit acute neutrophil mobilization from the bone marrow, suppressed accumulation of the immature granulocytic subpopulation, and a skewed long-term mobilization response. Furthermore, STAT3-deficient neutrophils are dramatically impaired in CXCR2-dependent chemotactic activity and demonstrate deregulated actin polymerization responses. These functions of STAT3 are independent of SOCS3, indicating that separate STAT3 pathways are essential for the actions of G-CSF in vivo.
Materials and methods
Bone marrow STAT3-deficient mice; myeloid SOCS3-deficient mice; G-CSF administration in vivo
STAT3-deficient mice were generated by breeding TIE2cre mice with floxed STAT3 mice.24,25 Genomic DNA isolated from tails of 3-week-old mice was used for genotyping by polymerase chain reaction (PCR) with primers described previously to detect wild-type, floxed, and deleted STAT3 alleles26 or the TIE2cre transgene.24 Deletion of STAT3 was maximized by generating animals carrying 1 floxed STAT3 allele and 1 deleted STAT3 allele (TIE2creSTAT3Δ/flox; termed herein as STAT3-deficient or KO), which is possible due to TIE2cre expression in the female germ line.24 Control animals contained 2 intact STAT3 alleles (eg, STAT3WT/WT, STAT3floxed/floxed, or STAT3WT/floxed genotypes) and lacked the TIE2cre transgene. In some cases, STAT3WT/WT mice containing the TIE2cre transgene were evaluated; these experiments ruled out contribution of transgene expression to the observed phenotypes (data not shown). Bone marrow samples from each animal were tested for STAT3 protein expression to confirm efficient deletion. Experiments were performed on mice 3 to 8 weeks of age. Mice were housed in a specific pathogen-free (SPF) facility and used in compliance with the Institutional Animal Care and Use Committee (IACUC) guidelines at the University of Texas M. D. Anderson Cancer Center (UT MDACC).
Myeloid SOCS3-deficient mice were generated by breeding LysMcre mice with floxed SOCS3 mice.26,27 Genomic DNA isolated from tails of 3-week-old mice was used for genotyping by PCR as described previously.26,27 SOCS3 deletion was verified in G-CSF–stimulated bone marrow granulocytes by quantitative real-time PCR. Total RNA was reverse transcribed as described.28 SOCS3 expression was measured using the following primers (RL86: 5′-CGGAGATTTCGCTTCGGGAC-3′; RL87: 5′-AACTTGCTGTGGGTGACCATGG-3′) and a FAM-labeled probe (5′-AGCTCCCCGGGATGCGGT-3′). Expression of β-actin was determined with primers and real-time PCR probes as described.29 Complementary DNA samples were analyzed in duplicate, and SOCS3 expression was normalized to β-actin as described previously.29 Mice were housed in an SPF facility and used in compliance with the IACUC guidelines at St Jude Children's Research Hospital.
Recombinant human G-CSF (Amgen, Thousand Oaks, CA) was diluted in sterile PBS containing 1% low-endotoxin BSA (Sigma, St Louis, MO). G-CSF was administered by subcutaneous injection using 250 μg/kg for a single dose, 125 μg/kg delivered every 12 hours for 5 days, or once daily at 250 μg/kg for 5 days, as indicated. Control mice received an equivalent volume of 1% BSA in PBS in parallel treatments.
Peripheral blood, bone marrow, and spleen isolation; blood counts; estimation of total hematopoietic neutrophil levels; G-CSF stimulation; and immunoblot analysis
Peripheral blood was collected in ethylenediaminetetraacetic acid (EDTA)–coated tubes following retro-orbital puncture (Becton Dickinson, Franklin Lakes, NJ). Complete blood counts were determined with a CELL-DYN 3700 automated hematology analyzer (Abbott Laboratories, Abbott Park, IL) through the UT MDACC Department of Veterinary Medicine. For additional analyses, blood samples were mixed with an equivalent volume of 2% dextran sulfate (Sigma) in PBS and incubated 20 minutes at room temperature to allow red blood cells to settle. The leukocyte-containing supernatant was removed and remaining red blood cells lysed by the addition of 5 mL buffer containing150 mM NH4Cl, 1 mM KHCO3, and 0.1 mM Na2 EDTA. Cells were washed and resuspended in PBS containing 2% FCS (PBS/FCS) for further use. Bone marrow cells were harvested in DME containing 10% FCS and 1% antibiotic-antimycotic (Gibco, Grand Island, NY) (DME/FCS). Spleens were harvested and homogenized in DME/FCS. Bone marrow and spleen cell suspensions were subjected to red blood lysis washed in DME/FCS, and used as indicated.
To calculate the total number of neutrophils present in the entire bone marrow and peripheral blood of an individual mouse, we used previously determined values of 0.075 mL/g for whole blood volume and a femur equivalent to 6% of the total bone marrow.30-32 Absolute levels of Gr-1+/CD11b+ cells were determined for total bone marrow, total peripheral blood, or spleen. Total hematopoietic neutrophil levels for each mouse were calculated by determining the sum of blood, bone marrow, and splenic neutrophils.
To test STAT3 expression and activation, bone marrow cells were stimulated with G-CSF (100 ng/mL) for 30 minutes at 37°C or left untreated. Whole cell lysates were subjected to immunoblot analysis with antibodies specific for tyrosine-phosphorylated STAT3 (Cell Signaling Technology, Danvers, MA), total STAT3 (Santa Cruz Biotechnology, Santa Cruz, CA), or STAT5 (Santa Cruz Biotechnology) as described previously.33
Antibody staining and flow cytometry; fluorescence-activated cell sorting (FACS); cytospins
For CXCR2 staining, cells were resuspended in 0.5% BSA (low endotoxin) in PBS and labeled with fluorescein isothiocyanate (FITC)–conjugated anti–Gr-1 (BD Pharmingen, San Jose, CA) and phycoerythrin (PE)–conjugated anti-CXCR2 (R&D Systems, Minneapolis, MN) or the appropriate isotype control antibodies and analyzed by flow cytometry using a Beckman Coulter (Miami, FL) Epics XL-MCL machine. For Gr-1/CD11b staining, cells were resuspended in PBS/2% FCS and incubated with FITC-conjugated anti–Gr-1 and PE-conjugated anti-CD11b (PharMingen) or isotype control antibodies. Samples from STAT3-deficient and control mice were analyzed on a BD FACSAria machine; samples from SOCS3-deficient and control mice were analyzed on a FACSCalibur machine. The bone marrow Gr-1lo/CD11b+ and Gr-1hi/CD11b+ populations were collected by fluorescence-activated cell sorting (FACS) (BD FACSAria) in DME/10% FCS. Cytospins were performed with 105 cells using electrostatic slides (Fisher Scientific, Hampton, NH) with a Cytospin 3 (Shandon, Waltham, MA). Cells were stained with the Hema3 staining kit according to the manufacturer's instructions (Fisher Diagnostics, Middletown, VA).
Isolation of bone marrow neutrophils; ex vivo neutrophil differentiation
Bone marrow neutrophils were isolated by positive selection using an AutoMacs column after labeling with FITC-conjugated anti–Gr-1 (PharMingen) and anti-FITC magnetic beads (Miltenyi Biotec, Auburn, CA). The purity of the neutrophil population was confirmed by flow cytometry and cytospin analyses, routinely demonstrating more than 90% Gr-1+/Mac-1+ cells with band/segmented morphology (data not shown).
To test cell autonomous functions, neutrophils were derived ex vivo from lineage-negative (lin–) bone marrow progenitor cells. Lin– cells were isolated by negative selection after staining with anti–Ly-1, anti-TER119, anti-B220, anti–Gr-1, anti–Mac-1, and anti–7-4 (StemCell Technologies, Vancouver, BC, Canada) and magnetic bead separation (AutoMacs). Lin– cells were then cultured in DME/FCS containing 100 ng/mL G-CSF and 10 ng/mL SCF (R&D Systems) for 12 days in a humidified CO2 incubator. Morphologic and flow cytometric analyses were performed at day 12, confirming the presence of band/segmented neutrophils and more than 85% Gr-1+/Mac-1+ cells in the cultures (data not shown).
Chemotaxis and actin polymerization assays
Neutrophils were resuspended in 1 × Hanks balanced salt solution containing calcium and magnesium (Gibco) and 0.1% BSA and plated in 3-μm Transwells (Fisher Scientific; 1 × 106 cells per Transwell) in the absence or presence of the indicated chemokine in the lower chamber (250 ng/mL MIP-2, R&D Systems; 1 μg/mL KC, Peprotech, Rocky Hill, NJ; 0.1 mM fMLP, Sigma). Transwells were incubated for 3 hours at 37°C in a humidified CO2 incubator; the number of cells that migrated through the Transwell was determined by enumeration. Cell viability was more than 90% as judged by trypan blue exclusion. To measure F-actin content, bone marrow cells were labeled with PE-conjugated anti–Gr-1 (eBioscience, San Diego, CA) as described in “Antibody staining and flow cytometry.” Cells were treated with MIP-2 (250 ng/mL) for 15, 30, 60, 120, 300, or 600 seconds or left untreated and then simultaneously fixed and stained with FITC-conjugated phalloidin (Molecular Probes, Eugene, OR) as described previously.34 The mean fluorescence intensity (MnI) of the phalloidin-FITC signal was determined for the Gr-1–PE–positive population by flow cytometry. The relative F-actin content (%F-actin) of each sample was determined by normalizing the sample MnI to the value for the untreated control animal.
Statistical analysis
Data are presented as mean values ± standard error mean (SEM); P values were calculated using an unpaired 2-tailed Student t test.
Results
Conditional deletion of STAT3 in the hematopoietic system
Bone marrow STAT3 deletion was accomplished by breeding TIE2cre mice with floxed STAT3 mice that contain loxP sites flanking the critical tyrosine (Y705) required for STAT3 activation.24,25 Genotypes were identified by PCR and confirmed by STAT3 immunoblot of bone marrow whole cell lysates. As predicted, cre-mediated deletion of STAT3 was highly efficient and, consistent with this, no phosphorylated STAT3 was detected following G-CSF stimulation. Moreover, full-length STAT3 protein was not detectable (Figure 1A-B). A very low level of a truncated form of STAT3 was observed, although this was not G-CSF responsive (Figure 1B). To ensure uniformity and rigorous examination of STAT3 function, individual bone marrow samples were examined by STAT3 immunoblotting, and only mice demonstrating efficient STAT3 deletion, as judged by the lack of STAT3 protein, were used for the studies described herein.
Figure 1.

Conditional deletion of STAT3 in bone marrow. (A) Genomic tail DNA was analyzed by PCR using primers that detect wild-type (w), floxed (f), and deleted (Δ) STAT3 alleles or the TIE2cre transgene (+). (B) Bone marrow cells from wild-type (WT) or STAT3-deficient (KO) mice were stimulated with G-CSF (100 ng/mL) for 30 minutes or left untreated, as indicated. Immunoblots of whole cell lysates were probed with antibodies specific for phospho-STAT3, total STAT3, or total STAT5. (C) Peripheral blood samples from WT or KO mice were stained with Gr-1 and CD11b antibodies conjugated to FITC and PE, respectively, and analyzed by flow cytometry. The percentage of Gr-1+/CD11b+ cells in individual samples was plotted; bars indicate the mean for each group (P < .001 comparing WT with KO).
STAT3-deficient mice showed elevated white blood cell counts, with increased levels of circulating neutrophils, monocytes, basophils, and eosinophils. Absolute red blood cell counts were normal, although STAT3-deficient mice have reduced hematocrit and hemoglobin levels (Table 1; Figure 1C). The peripheral neutrophilia observed in STAT3-deficient mice is consistent with a negative role for STAT3 in basal granulopoiesis.15-17
Table 1.
Hematologic parameters and WBC differential count of STAT3-deficient mice
| WT | KO | |
|---|---|---|
| Hematologic parameters | ||
| Hemoglobin level, g/L | 159.7 ± 6.9 | 123.7 ± 6.6* |
| Hematocrit | .4713 ± .0208 | .3726 ± .0216* |
| WBC count, × 109/L | 8.93 ± 1.04 | 14.53 ± 1.55† |
| RBC count, × 1012/L | 10.14 ± 0.47 | 8.77 ± 0.53 |
| Platelet count, × 109/L | 996 ± 53 | 1029 ± 107 |
| WBC differential count | ||
| Neutrophils, × 109/L | 0.96 ± 0.08 | 3.49 ± 0.60* |
| Monocytes, × 109/L | 0.24 ± 0.10 | 1.53 ± 0.36* |
| Basophils, × 109/L | 0.20 ± 0.04 | 0.78 ± 0.24† |
| Lymphocytes, × 109/L | 7.42 ± 1.04 | 8.51 ± 1.34 |
| Eosinophils, × 109/L | 0.11 ± 0.02 | 0.23 ± 0.04† |
Peripheral blood analysis from STAT3-deficient mice (KO) and wild-type (WT) littermates. Results represent mean ± SEM (n = 7 for WT and KO).
WBC indicates white blood cell; RBC, red blood cell.
P < .01 compared with WT mice.
P < .05 compared with WT mice.
STAT3 controls G-CSF–dependent expansion of the immature granulocytic population in vivo
A principal function of G-CSF is to stimulate the growth of neutrophil precursors during emergency granulopoiesis.5 To examine the role of STAT3 in this response, mice were given a single dose of G-CSF, treated twice daily for 5 days, or were injected with BSA-containing buffer alone. Bone marrow samples were evaluated for immature and mature granulocytes by Gr-1/CD11b staining and flow cytometry. In wild-type mice, Gr-1 expression increases during granulocytic maturation, and Gr-1lo/CD11b+ and Gr-1hi/CD11b+ subsets define immature and mature neutrophil populations, respectively.35 Wild-type mice that received BSA alone have a proportion of immature and mature bone marrow granulocytes similar to that of untreated mice, with an approximate 1:2 ratio of Gr-1lo/CD11b+ to Gr-1hi/CD11b+ cells (Figure 2A-B and data not shown), demonstrating that BSA administration does not affect the granulocytic population. STAT3 deletion does not alter this distribution under basal conditions (eg, in untreated or BSA-treated animals) (Figure 2A-B and data not shown), in agreement with previous reports that found STAT3 was dispensable for maintaining granulocytic progenitor levels under steady-state conditions.15,17 Strikingly, however, STAT3-deficient mice exhibited a dramatically altered bone marrow response to G-CSF. By 24 hours following a single dose of G-CSF, wild-type mice demonstrated a 2-fold induction in the immature granulocytic compartment (eg, Gr-1lo/CD11b+ cells) and a concomitant drop in mature neutrophils in the bone marrow (Figure 2A). By contrast, STAT3-deficient mice failed to induce the immature granulocytic population and did not down-regulate mature Gr-1hi/CD11b+ bone marrow neutrophil levels (Figure 2A).
Figure 2.
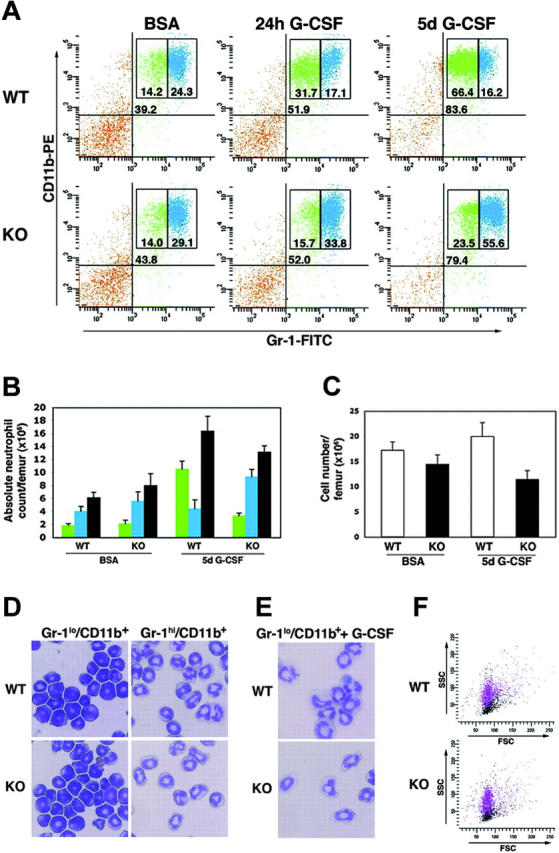
STAT3-deficient bone marrow granulocytes have an atypical response to G-CSF in vivo. (A-C) Wild-type (WT) or STAT3-deficient (KO) mice were treated with a single dose of G-CSF (250 μg/kg), twice daily doses of G-CSF for 5 days (125 μg/kg every 12 hours), or BSA-containing buffer. Bone marrow cells were collected 24 hours following the single G-CSF dose or 3 hours following the last G-CSF dose in the 5-day treatment regimen. Cells were stained with anti–Gr-1 and anti-CD11b antibodies conjugated to FITC and PE, respectively. (A) Samples were analyzed by flow cytometry, and the percentage of total Gr-1+/CD11b+ cells (top right quadrant), Gr-1lo/CD11b+ cells (green), and Gr-1hi/CD11b+ cells (blue) is indicated. Data are representative of 3 independent experiments. (B) Absolute levels of Gr-1lo/CD11b+ cells (green), Gr-1hi/CD11b+ cells (blue), or total Gr-1+/CD11b+ cells (gray) are shown (n = 4 for WT and KO for each condition). Error bars represent SEM. (C) Mononuclear cell counts were determined for each femur (n = 5 for WT and KO for each condition). Error bars represent SEM. (D) The Gr-1lo/CD11b+ and Gr-1hi/CD11b+ populations were isolated from wild-type and STAT3-deficient bone marrow samples by FACS and used for cytospin preparations. Original magnification of the cytospins was × 400. Photos were taken using a Nikon Microphot-FX microscope (Nikon, Garden City, NJ) using a PlanApo 40×/0.95 numerical aperture objective and equipped with a 3-chip charged coupled device (CCD) color video camera (model DXC990; Sony, Tokyo, Japan). Digital images were captured using Optimas Image Analysis software (Media Cybernetics, Silver Spring, MD). Data are representative of 2 independent experiments. (E) Bone marrow Gr-1lo/CD11b+ cells from wild-type and STAT3-deficient mice were isolated by FACS and grown in G-CSF for 3 days ex vivo. Cytospin preparations of the differentiated cells are shown; original magnification was × 400, and images were captured as described for panel D. Data are representative of 2 independent experiments. (F) Forward scatter (FSC) and side scatter (SSC) analysis of Gr-1lo/CD11b+ (black) and Gr-1hi/CD11b+ (purple) populations from bone marrow of WT and KO mice is shown. Data are representative of 5 independent experiments.
Long-term exposure to G-CSF also rendered a skewed response in the bone marrow of STAT3-deficient mice. Wild-type animals had a substantial induction (4- to 5-fold) in the frequency and absolute numbers of immature bone marrow granulocytes following 5 days of G-CSF treatment. In comparison, STAT3-deficient mice showed a less than 2-fold increase in the immature bone marrow Gr-1lo/CD11b+ compartment relative to basal levels (Figure 2A-B). Importantly, the low number of immature granulocytes in G-CSF–treated STAT3-deficient mice was not due to increased cell death within this population, as judged by propidium iodide uptake (data not shown). Moreover, the frequency and absolute numbers of mature neutrophils were increased in the bone marrow of STAT3-deficient animals following G-CSF treatment, while wild-type mice had a reduced proportion and similar absolute levels of mature Gr-1hi/CD11b+ cells relative to BSA-treated or untreated controls (Figure 2A-B and data not shown). Mice of both genotypes showed elevated bone marrow neutrophil counts following sustained exposure to G-CSF, yet the total marrow cell count was reduced in STAT3-deficient mice (Figures 2B-C).
To verify that the Gr-1 marker protein is regulated similarly in wild-type and STAT3-deficient mice, Gr-1lo/CD11b+ and Gr-1hi/CD11b+ subsets were isolated by FACS and subjected to cytospin analysis. More than 90% of Gr-1hi/CD11b+ cells from wild-type or STAT3-deficient bone marrow showed band/segmented morphology, demonstrating that this population consists primarily of mature neutrophils in both genotypes (Figure 2D). Importantly, Gr-1lo/CD11b+ cells sorted from wild-type and STAT3-deficient mice exhibited an immature morphology, as indicated by an increased nuclear-cytoplasmic ratio and a lack of band/segmented cells (Figure 2D). These results are consistent with previous analyses of Gr-1lo/CD11b+ cells from wild-type mice, which showed that this population was composed mainly of myelocytes and immature progenitors.35 To examine whether the immature subset showed similar developmental potential in wild-type and STAT3-deficient backgrounds, Gr-1lo/CD11b+ cells were sorted by FACS and cultured ex vivo in medium containing G-CSF for 3 days. These assays revealed that Gr-1lo/CD11b+ cells gave rise to cells with mature band/segmented neutrophil morphology, demonstrating comparable maturation of immature granulocytes from both genotypes (Figure 2E). Moreover, forward and side scatter analysis of bone marrow Gr-1+/CD11b+ cells from wild-type and STAT3-deficient mice demonstrated comparable granularity and size, with Gr-1hi/CD11b+ cells exhibiting increased granularity relative to the Gr-1lo/CD11b+ subset (Figure 2F). Thus, low and high Gr-1 cell-surface expression levels define immature and mature granulocytes, respectively, in both wild-type and STAT3-deficient mice. Collectively, therefore, our results show that STAT3-deficient mice have an altered bone marrow response to G-CSF, characterized by a failure to accumulate immature granulocytic cells, lower total bone marrow cellularity, and a slight increase in mature neutrophils relative to wild-type animals.
Role for STAT3 in the peripheral neutrophil response to G-CSF
To examine the effect of STAT3 deficiency on peripheral neutrophil responses to G-CSF, we treated mice with G-CSF twice daily for 5 days and examined circulating and splenic neutrophil populations. Wild-type mice showed a robust induction of peripheral blood neutrophils, as evidenced by an increase in the percentage of circulating Gr-1+/CD11b+ cells as well as absolute neutrophil counts (Figure 3A-B). STAT3-deficient mice also showed significant induction of peripheral blood neutrophil levels following sustained exposure to G-CSF; however, there was a dramatic difference in the proportion of mature to immature granulocytes that accumulated relative to wild-type mice (Figures 3A-B). Wild-type mice had a substantial increase in circulating immature granulocytes (eg, Gr-1lo/CD11b+ cells) as anticipated4,35 (Figures 3A-B), consistent with the accumulation of this population in the bone marrow (Figure 2A-B). The frequency of mature peripheral blood neutrophils (eg, Gr-1hi/CD11b+ cells) was relatively unchanged in wild-type mice exposed to G-CSF, although absolute numbers increased (Figures 3A-B). By contrast, STAT3-deficient animals showed a poor induction of immature granulocytes in peripheral blood (Figures 3A-B), in agreement with the relative paucity of this subset in their bone marrow compartment (Figures 2A-B). Mature Gr-1hi/CD11b+ cells, however, accumulated in the peripheral blood of STAT3-deficient mice following sustained exposure to G-CSF (Figures 3A-B). Splenic neutrophil populations showed a similar trend, with wild-type mice exhibiting an elevated ratio of immature to mature neutrophils, while STAT3-deficient mice showed an increased ratio of mature to immature neutrophils following sustained exposure to G-CSF (Figure 3C). No significant differences were found in total spleen cellularity between wild-type and STAT3-deficient mice under basal conditions, and both genotypes showed a similar increase in spleen cell content following G-CSF treatment (Figure 3D).
Figure 3.
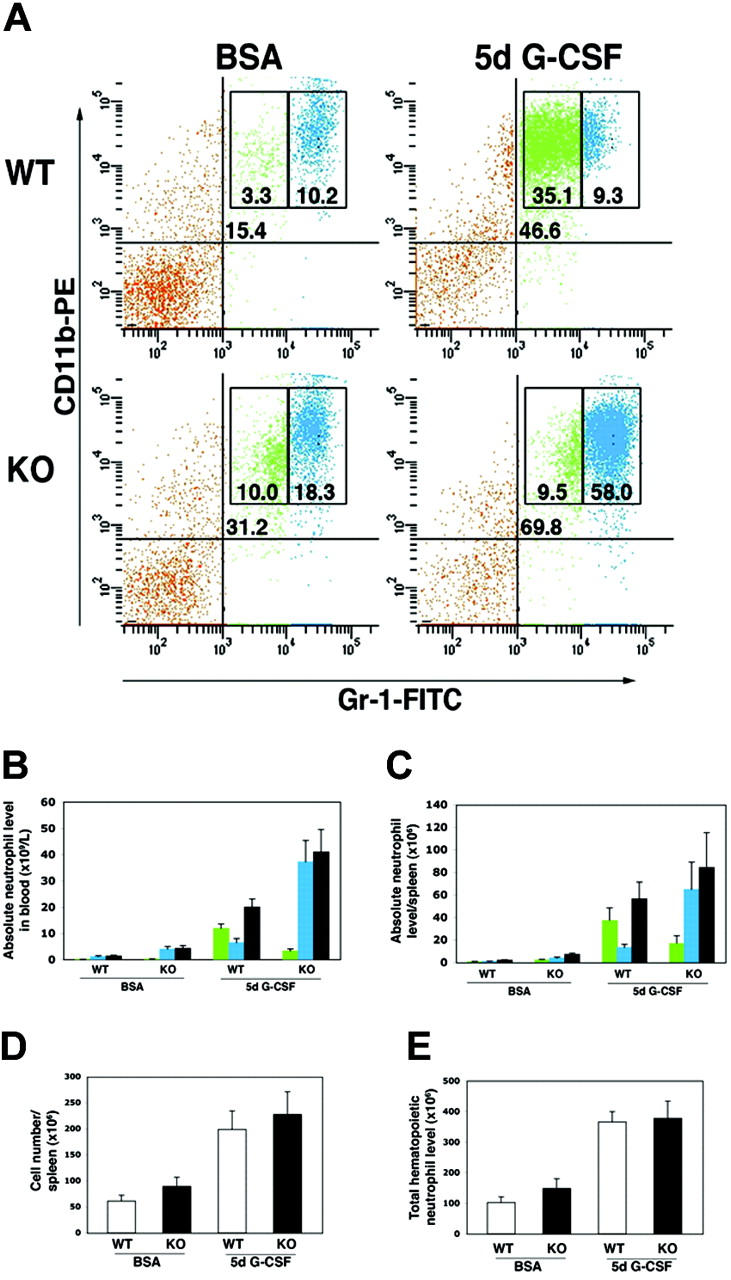
STAT3 is required to maintain a normal ratio of immature to mature neutrophils in peripheral blood and spleen. Wild-type (WT) and STAT3-deficient (KO) mice were injected at 12-hour intervals with G-CSF (125 μg/kg) or BSA-containing buffer for 5 days. Peripheral blood and spleen samples were collected 3 hours following the last injection and stained with anti–Gr-1 and anti-CD11b antibodies conjugated to FITC and PE, respectively. (A) Blood samples were analyzed by flow cytometry, and the percentage of total Gr-1+/CD11b+ cells (top right quadrant), Gr-1lo/CD11b+ cells (green), and Gr-1hi/CD11b+ cells (blue) is indicated. Because STAT3-deficient mice show variable neutrophilia, animals with similar basal levels of peripheral Gr-1+/CD11b+ cells were chosen for comparison. Results are representative of at least 2 independent experiments (n = 4 for WT and KO for each condition). (B-C) Absolute levels of Gr-1lo/CD11b+ cells (green), Gr-1hi/CD11b+ cells (blue), or total Gr-1+/CD11b+ cells (gray) in blood (B) or spleen (C) are shown (n = at least 4 for WT and KO for each condition). (D) Mononuclear cell counts were determined for individual spleens (n = 4 for WT and KO for each condition). (E) Total hematopoietic neutrophil levels were determined per mouse as described in “Materials and methods.” Average neutrophil numbers are shown (n = 4 for WT and KO for each condition). Error bars represent SEM in all panels.
Taken together, our data show that STAT3 is required for induction of immature neutrophils in the bone marrow and the peripheral hematopoietic compartments in response to G-CSF. STAT3 also controls the level of mature neutrophils that accumulate in marrow, blood, and spleen following G-CSF treatment (Figures 3A-C). To ascertain whether these changes in immature and mature neutrophil numbers affected the total level of neutrophils in STAT3-deficient mice, the sum of absolute neutrophil numbers in whole bone marrow, blood, and spleen was determined for G-CSF– and BSA-treated mice. These data show that the total hematopoietic neutrophil content was increased upon G-CSF treatment but did not differ significantly between wild-type and STAT3-deficient mice in basal conditions or following G-CSF (Figure 3E).
SOCS3 is not required for induction of immature granulocytes in response to G-CSF
STAT3 function in the neutrophil lineage has been largely attributed to its control of SOCS3, a critical negative regulator of G-CSF.15,18 To examine whether SOCS3 was necessary for the STAT3-dependent functions that we found in emergency granulopoiesis, mice with myeloid-specific SOCS3 deletion were treated with G-CSF for 5 days, and bone marrow and peripheral blood Gr-1 subsets were analyzed by flow cytometry. Wild-type and myeloid SOCS3-deficient mice showed a robust accumulation of immature Gr-1lo/CD11b+ cells in the bone marrow and peripheral blood, with no apparent differences between the genotypes (Figure 4). Therefore, SOCS3 is dispensable for G-CSF–dependent induction of immature granulocytes, indicating the existence of separate STAT3-responsive pathways in the neutrophil lineage.
Figure 4.

SOCS3 is not required for G-CSF–induced expansion or mobilization of immature granulocytes. Wild-type (WT) or myeloid SOCS3-deficient (SOCS3 KO) mice were treated daily with G-CSF (250 μg/kg/d) or BSA-containing buffer for 5 days. Bone marrow (A) or peripheral blood (B) cells were collected 3 hours following the last G-CSF dose. Cells were stained with anti–Gr-1 and anti-CD11b antibodies conjugated to FITC and PE, respectively. Samples were analyzed by flow cytometry, and the percentage of total Gr-1+/CD11b+ cells (top right quadrant), Gr-1lo/CD11b+ cells (green), and Gr-1hi/CD11b+ cells (blue) is indicated. Results of a representative experiment are shown.
Role for STAT3 in neutrophil mobilization by G-CSF
The bone marrow contains a reserve pool of mature neutrophils that can be rapidly mobilized by G-CSF during infection and stress conditions of emergency granulopoiesis.36,37 This process requires neutrophil migration from sites of attachment on the bone marrow stroma, across sinusoidal endothelial cells, and into the peripheral circulation. G-CSF treatment of STAT3-deficient mice failed to down-regulate mature neutrophil levels in bone marrow by 24 hours, in contrast to its effect in wild-type animals (Figure 2A), suggesting that the acute neutrophil mobilization response to G-CSF may require STAT3. To test the role of STAT3, G-CSF was administered to wild-type and STAT3-deficient mice by subcutaneous injection, and circulating neutrophil levels were examined 24 hours after treatment. This enabled us to monitor the acute mobilization response while minimizing the contribution of G-CSF–induced nascent granulocyte production to circulating neutrophil levels.4,32 As expected, wild-type mice showed a rapid induction of peripheral neutrophil levels in response to G-CSF, with a 2- to 3-fold increase in their frequency and absolute numbers in the blood (Figure 5A-B). STAT3-deficient mice were refractory to this treatment, as evidenced by their failure to up-regulate circulating neutrophil levels by 24 hours (Figures 5A-B). Thus, the acute neutrophil mobilization response elicited by G-CSF depends on expression of STAT3 in the bone marrow.
Figure 5.

STAT3-deficient mice have impaired acute neutrophil mobilization in response to G-CSF. (A) Peripheral blood samples were analyzed by flow cytometry following red blood cell lysis to determine the percentage of Gr-1+/CD11b+ cells present in untreated mice or mice that received G-CSF (250 μg/kg) 24 hours prior to analysis. The amount of Gr-1+/CD11b+ cells in G-CSF–treated samples was normalized to the amount in untreated controls of the appropriate genotype to provide the relative level of Gr-1+/CD11b+ cells in the blood following G-CSF treatment. Relative Gr-1+/CD11b+ levels for wild-type (WT, □) and STAT3-deficient (KO, ▪) mice are shown (n = 6 for WT, n = 5 for KO) (P = .003 comparing WT with KO). (B) Blood samples were subjected to automated counting to determine total neutrophil numbers in untreated mice or mice that received G-CSF (250 μg/kg) 24 hours prior to analysis. Neutrophil amounts in G-CSF–treated samples were normalized to amounts in untreated controls of the appropriate genotype to determine the relative induction by G-CSF. Relative neutrophil levels for WT (□) and KO (▪) are shown (n = 6 for WT, n = 5 for KO) (P = .002 comparing WT with KO). Error bars represent SEM in both panels.
STAT3-deficient neutrophils show impaired chemotaxis toward CXCR2 ligands
G-CSFR regulates specific functional responses of mature neutrophils, including neutrophil chemotaxis and adhesion capabilities.6 We had previously observed a connection between STAT3 activation and enhanced function of myeloid adhesion molecules in 32D cells, suggesting that STAT3 may control pathways central to cell migration.33 We examined neutrophil chemotaxis in Transwell assays as a method to test the migratory capabilities of STAT3-deficient cells. Strikingly, we found that neutrophils isolated from the bone marrow of STAT3-deficient mice demonstrated a severely impaired response to the potent neutrophil chemoattractant MIP-2 relative to their wild-type counterparts (Figure 6A).
Figure 6.
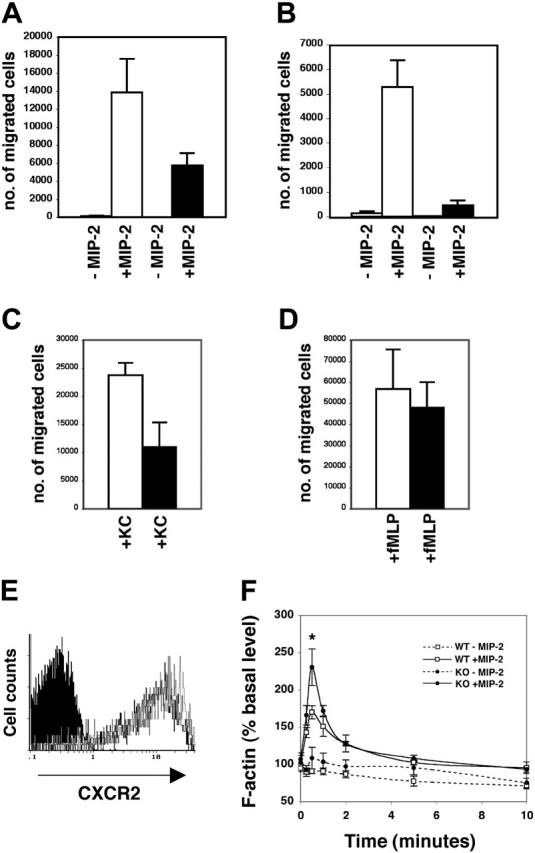
STAT3-deficient neutrophils have impaired CXCR2-mediated chemotaxis. (A) Bone marrow neutrophils from wild-type (WT, □) or STAT3-deficient (KO, ▪) mice were examined for MIP-2–dependent chemotaxis in migration assays with 3-μm Transwells (n = 6 per genotype) (P = .065 comparing WT with KO). Average values from at least 3 independent experiments are shown. (B) Neutrophils were derived from lin– progenitors in G-CSF–containing medium ex vivo. At day 12, neutrophils were tested for MIP-2–dependent chemotaxis (n = 5 for WT, □;n = 4 for KO, ▪) (P = .006 comparing WT with KO). Average values from at least 3 independent experiments are shown. (C) Bone marrow neutrophils were tested for chemotaxis toward KC (n = 5 for WT, □;n = 4 for KO, ▪)(P = .028 comparing WT with KO). Average values from at least 3 independent experiments are shown. (D) Chemotaxis toward fMLP was tested with bone marrow neutrophils as indicated (WT, □; KO, ▪; n = 4 per genotype) (P = .710 comparing WT with KO). Average values from at least 3 independent experiments are shown. (E) Bone marrow cells from wild-type or STAT3-deficient mice were stained with antibodies for Gr-1 and CXCR2 and examined by flow cytometry. The histogram shows CXCR2 cell-surface expression in the Gr-1+ population from WT (open gray) and KO (open black) mice relative to isotype control (solid black). Results are representative of 3 independent experiments. (F) Bone marrow cells were stained with PE-conjugated Gr-1 antibody and then stimulated with MIP-2 or left untreated for the times indicated. Cells were fixed and stained with phalloidin-FITC and analyzed by flow cytometry. Relative F-actin levels were determined as described in “Chemotaxis and actin polymerization assays” and plotted against MIP-2 treatment time points (n = 5 per genotype) (P = .049, F-actin at 30 seconds comparing WT with KO). Average values from 4 independent experiments are shown. Error bars represent SEM in all panels.
To evaluate a potential cell autonomous role for STAT3, neutrophils were derived from lin– hematopoietic progenitor cells (HPCs) in an ex vivo culture system that depends exclusively on G-CSF. Wild-type neutrophils derived ex vivo showed MIP-2–dependent migration, indicating that G-CSF–driven differentiation faithfully replicates the developmental pathway necessary to acquire chemotactic function. By contrast, STAT3-deficient neutrophils derived ex vivo showed a severe block in MIP-2–dependent chemotaxis (Figure 6B). Both freshly isolated bone marrow neutrophils and ex vivo–derived neutrophils remained viable at the end of the chemotaxis assay, as judged by trypan blue staining, ruling out the possibility that changes in cell survival affect chemotactic function. Collectively, the data show that STAT3 serves a cell-autonomous role in neutrophil chemotaxis toward MIP-2. This function of STAT3 is independent of its control of SOCS3 expression because neutrophils from myeloid SOCS3-deficient mice demonstrate normal chemotactic activity in response to MIP-2 (data not shown).
To examine the specificity of the chemotactic defect, we tested the response of neutrophils to the chemoattractants KC and fMLP. Like MIP-2, KC signals via the CXCR2 chemokine receptor while fMLP functions through a distinct 7-transmembrane receptor.38-41 STAT3-deficient neutrophils are severely impaired in KC-dependent chemotaxis, confirming defective migration in response to signals elicited by the CXCR2 receptor (Figure 6C). Interestingly, STAT3-deficient bone marrow neutrophils migrate toward fMLP with an activity similar to wild-type controls (Figure 6D). These results show that STAT3 has a selective function in the CXCR2 response and indicate that the high degree of specificity in neutrophil chemoattractant signaling pathways is regulated in part by STAT3.
Wild-type and STAT3-deficient neutrophils demonstrated similar levels of cell-surface CXCR2 receptor and CXCR2 mRNA (Figure 6E and data not shown), indicating that expression of the MIP-2/KC receptor is independent of STAT3. To evaluate CXCR2 signaling, we tested MIP-2–dependent actin reorganization because this response is essential for directed cellular migration.42 F-actin content was measured in neutrophils by staining MIP-2–stimulated and untreated bone marrow cells with Gr-1 antibody and phalloidin-FITC. These assays revealed that wild-type and STAT3-deficient neutrophils responded to MIP-2 by a rapid burst of actin polymerization, followed by depolymerization to basal levels (Figure 6F). The kinetics of actin polymerization and depolymerization were similar between wild-type and STAT3-deficient neutrophils, indicating that the global signaling response to MIP-2 is intact in the STAT3-deficient cells. Interestingly, however, STAT3-deficient neutrophils showed an elevated level of polymerized actin at the peak of stimulation (30 seconds) (Figure 6F), which suggests a hyperactivated polymerization signal. These findings indicate that STAT3 controls the magnitude of actin polymerization in response to CXCR2 signaling, which may result in changes in cell rigidity or downstream functions required for chemotaxis.
Discussion
We find that STAT3 has a fundamental role in the emergency granulopoiesis response and in the function of neutrophils produced in steady-state hematopoiesis. STAT3 is necessary for acute neutrophil mobilization by G-CSF, accumulation of immature granulocytes, and maintenance of normal ratios of immature to mature neutrophils in bone marrow, peripheral blood, and spleen during G-CSF–driven emergency granulopoiesis. Moreover, STAT3 is essential for neutrophil chemotaxis toward natural CXCR2 ligands and control of the magnitude of chemoattractant-induced actin polymerization. Previous studies have shown that STAT3 is a negative regulator of steady-state granulopoiesis. Our data demonstrate that STAT3 also has important positive regulatory roles in the granulocytic lineage.
Accumulation of mature neutrophils in STAT3-deficient mice is attributed to a negative regulatory loop initiated by STAT3, which stimulates expression of SOCS3, a crucial feedback inhibitor of G-CSF.15,18 We found that SOCS3 was dispensable, however, for the induction of immature granulocytes in response to G-CSF and for CXCR2-mediated neutrophil chemotaxis. These data collectively argue that STAT3 activates SOCS3-independent target pathways in neutrophils. Indeed, bone marrow conditional STAT3 and SOCS3 knockout mice have important differences in phenotype, including their ability to tolerate high doses of G-CSF administered over several days (this study, Lee et al,15 Kamezaki et al,17 and Croker et al18). Moreover, SOCS3 deficiency does not affect neutrophil function, while deletion of STAT3 impairs chemotaxis and respiratory burst activities.16,18 The identification of STAT3 target pathways that are independent of SOCS3 is of considerable interest for understanding the molecular mechanisms of G-CSF and STAT3 in granulocytes.
Expansion of the immature granulocytic population is an important component of the emergency granulopoiesis response, providing precursors to support a sustained increase in the circulating neutrophil pool. This response is exploited clinically with the use of G-CSF to treat neutropenia, particularly in cancer patients undergoing chemotherapy. STAT3-deficient mice showed a marked impairment in G-CSF–stimulated accumulation of immature granulocytes in bone marrow, indicating that STAT3 controls their response to G-CSF. Mature neutrophils were increased in STAT3-deficient mice during G-CSF treatment, resulting in an atypical G-CSF response characterized by an elevated ratio of mature to immature neutrophils. This skewed ratio was also found in peripheral blood and spleen, thus indicating that immature granulocytes are underrepresented in STAT3-deficient mice, rather than selectively sequestered in a peripheral hematopoietic compartment.
The reduced number of immature granulocytes and elevated level of mature neutrophils in STAT3-deficient mice following G-CSF treatment raised the question of whether total neutrophil output was affected by STAT3 deletion. Estimation of absolute neutrophil numbers in total bone marrow, peripheral blood, and spleen suggests that STAT3 has little impact on overall neutrophil production during steady-state or emergency granulopoiesis (Figure 3E). Rather, a major function for STAT3 is to regulate the ratio of immature to mature neutrophils generated in response to G-CSF. The negative function ascribed to STAT3 is likely the principal cause of increased mature neutrophil levels, by enhancing their survival and/or proliferation of an immediate precursor.15,18 This negative function, however, may be offset by a requirement for STAT3 to maintain the level or growth responsiveness of a granulocytic progenitor compartment, thus leading to impaired accumulation of immature granulocytes during G-CSF–driven granulopoiesis. STAT3 controls numerous genes that regulate cell growth in other tissues43; these pathways may stimulate granulocytic progenitors to proliferate during an emergency response to G-CSF. Alternatively, STAT3-deficient granulocytes may bypass the immature Gr-1lo compartment or mature at a faster rate through this developmental stage during a G-CSF response. Nonetheless, the altered response of STAT3-deficient granulocytes suggests that they may fail to progress through essential checkpoints during development, which could contribute to defects in mobilization and chemotaxis.
Signals through the G-CSFR are essential for steady-state and acute neutrophil release from the bone marrow.32 Acute neutrophil mobilization was attenuated in STAT3-deficient mice, as evidenced by their failure to down-regulate the mature neutrophil compartment in the bone marrow and induce peripheral neutrophil levels 24 hours after G-CSF administration. Under steady-state conditions or following prolonged exposure to G-CSF, however, peripheral neutrophil accumulation was observed in STAT3-deficient mice. Thus, while bone marrow expression of STAT3 is essential for G-CSF–stimulated acute neutrophil mobilization, it is not necessary for all neutrophil mobilization functions. Recent studies have shown that basal neutrophil release from the bone marrow is controlled by adhesion mechanisms that are distinct from acute mobilization responses,44 raising the possibility that STAT3 has a specific role in the acute release pathway.
STAT proteins have been linked with cellular adhesion and migration functions previously.22,33,45-47 The impaired chemotaxis of STAT3-deficient neutrophils was accompanied by an enhanced actin polymerization response to MIP-2, indicating that STAT3 operates in pathways that mediate CXCR2 signaling to the cytoskeleton. Interestingly, STAT3 is necessary for CXCR2-dependent migration but not chemotaxis toward fMLP. This indicates a previously unrecognized potential: CXCR2 and the fMLP receptor may have distinct signaling requirements despite their similarity as G protein–coupled receptors, with a selective requirement for STAT3 in CXCR2 responses.
In summary, our work provides direct evidence for a positive role for STAT3 in regulating essential aspects of G-CSF–driven emergency granulopoiesis and neutrophil function. This adds to the previously described negative function for STAT3 in controlling mature neutrophil levels. Future studies to elucidate STAT3 target genes in neutrophils and their progenitors are necessary to define the mechanisms that underlie the regulatory activities of STAT3 and establish how these pathways participate in granulopoiesis, which may aid in developing approaches to manage this essential blood cell lineage.
Authorship
A.D.P. and S.S.W. designed the study, designed experiments, performed experiments, analyzed data, and wrote the paper; P.J.M., A.M.S., and K.C.E.K. designed and performed experiments and analyzed data from myeloid SOCS3-deficient mice; and D.C.L., F.L., M.A.G., J.W.S., D.M.J., and L.Z. contributed to experiment design and analysis of results and performed experiments.
The authors declare no competing financial interests.
Acknowledgments
We are grateful to Drs Pandelakis Koni and Kiyoshi Takeda for access to TIE2cre and floxed STAT3 mice, respectively, and to Drs Doug Hilton and Warren Alexander for the gift of LysMcre+SOCS3flox/flox mice. We thank Drs Ben Kile, Doug Hilton, Brad McIntyre, and Miles Wilkinson for many stimulating and insightful discussions. We thank members of the McIntyre and Wilkinson laboratories for providing advice on numerous aspects of this project, Karen Ramirez and David He for their outstanding assistance with flow cytometry, and Carol Oborn for help with microscopy and image acquisition. We thank Dr Morgan McLemore for advice on in vitro neutrophil differentiation and Jennifer Carter and Hoainam Nguyen-Jackson for helpful discussion.
This work was supported by a National Institutes of Health (NIH) predoctoral training grant in Cancer Immunology (T32-CA-09598-16) and an American Legion Auxiliary Award (A.D.P.) as well as by grants from the NIH (CA77447), American Heart Association (AHA) Texas Affiliate (0255899Y, 0455143Y), Gillson Longenbaugh Foundation, and the MDACC Institutional Grants Program (S.S.W.).
Prepublished online as Blood First Edition Paper, August 3, 2006; DOI 10.1182/blood-2006-02-003012.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
References
- 1.Nicola NA, Metcalf D, Johnson GR, Burgess AW. Separation of functionally distinct human granulocyte-macrophage colony stimulating factors. Blood. 1979;54: 614-627. [PubMed] [Google Scholar]
- 2.Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity. 1996;5: 491-501. [DOI] [PubMed] [Google Scholar]
- 3.Lieschke GJ, Grail D, Hodgson G, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84: 1737-1746. [PubMed] [Google Scholar]
- 4.Basu S, Hodgson G, Katz M, Dunn AR. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood. 2002;100: 854-861. [DOI] [PubMed] [Google Scholar]
- 5.Lord BI, Molineux G, Podja Z, Souza LM, Mermod JJ, Dexter TM. Myeloid cell kinetics in mice treated with recombinant interleukin-3, granulocyte colony-stimulating factor (CSF), or granulocyte-macrophage CSF in vivo. Blood. 1991;77: 2154-2159. [PubMed] [Google Scholar]
- 6.Betsuyaku T, Liu F, Senior RM, et al. A functional granulocyte colony-stimulating factor receptor is required for normal chemoattractant-induced neutrophil activation. J Clin Invest. 1999;103: 825-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Semerad CL, Poursine-Laurent J, Liu F, Link DC. A role for G-CSF receptor signaling in the regulation of hematopoietic cell function but not lineage commitment or differentiation. Immunity. 1999;11: 153-161. [DOI] [PubMed] [Google Scholar]
- 8.O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109: S121-S131. [DOI] [PubMed] [Google Scholar]
- 9.Snow JW, Abraham N, Ma MC, Abbey NW, Herndier B, Goldsmith MA. STAT5 promotes multilineage hematolymphoid development in vivo through effects on early hematopoietic progenitor cells. Blood. 2002;99: 95-101. [DOI] [PubMed] [Google Scholar]
- 10.Moriggl R, Sexl V, Kenner L, et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell. 2005;7: 87-99. [DOI] [PubMed] [Google Scholar]
- 11.Bunting KD, Bradley HL, Hawley TS, Moriggl R, Sorrentino BP, Ihle JN. Reduced lymphomyeloid repopulating activity from adult bone marrow and fetal liver of mice lacking expression of STAT5. Blood. 2002;99: 479-487. [DOI] [PubMed] [Google Scholar]
- 12.Teglund S, McKay C, Schuetz E, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93: 841-850. [DOI] [PubMed] [Google Scholar]
- 13.Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84: 443-450. [DOI] [PubMed] [Google Scholar]
- 14.Meraz MA, White JM, Sheehan KC, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84: 431-442. [DOI] [PubMed] [Google Scholar]
- 15.Lee CK, Raz R, Gimeno R, et al. STAT3 is a negative regulator of granulopoiesis but is not required for G-CSF-dependent differentiation. Immunity. 2002;17: 63-72. [DOI] [PubMed] [Google Scholar]
- 16.Welte T, Zhang SS, Wang T, et al. STAT3 deletion during hematopoiesis causes Crohn's disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A. 2003;100: 1879-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamezaki K, Shimoda K, Numata A, et al. Roles of Stat3 and ERK in G-CSF signaling. Stem Cells. 2005;23: 252-263. [DOI] [PubMed] [Google Scholar]
- 18.Croker BA, Metcalf D, Robb L, et al. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity. 2004;20: 153-165. [DOI] [PubMed] [Google Scholar]
- 19.McLemore ML, Grewal S, Liu F, et al. STAT-3 activation is required for normal G-CSF-dependent proliferation and granulocytic differentiation. Immunity. 2001;14: 193-204. [DOI] [PubMed] [Google Scholar]
- 20.Hermans MH, Antonissen C, Ward AC, Mayen AE, Ploemacher RE, Touw IP. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med. 1999;189: 683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van de Geijn GJ, Gits J, Aarts LHJ, Heijmans-Antonissen C, Touw IP. G-CSF receptor truncations found in SCN/AML relieve SOCS3-controlled inhibition of STAT5 but leave suppression of STAT3 intact. Blood. 2004;104: 667-674. [DOI] [PubMed] [Google Scholar]
- 22.Panopoulos AP, Bartos D, Zhang L, Watowich SS. Control of myeloid-specific integrin αMβ2 (CD11b/CD18) expression by cytokines is regulated by Stat3-dependent activation of PU.1. J Biol Chem. 2002;277: 19001-19007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimozaki K, Nakajima K, Hirano T, Nagata S. Involvement of STAT3 in the granulocyte colony-stimulating factor-induced differentiation of myeloid cells. J Biol Chem. 1997;272: 25184-25189. [DOI] [PubMed] [Google Scholar]
- 24.Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med. 2001;193: 741-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998;161: 4652-4660. [PubMed] [Google Scholar]
- 26.Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10: 39-49. [DOI] [PubMed] [Google Scholar]
- 27.Croker BA, Krebs DL, Zhang JG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4: 540-545. [DOI] [PubMed] [Google Scholar]
- 28.Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J Immunol. 2002;168: 3402-3411. [DOI] [PubMed] [Google Scholar]
- 29.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169: 2253-2263. [DOI] [PubMed] [Google Scholar]
- 30.Chervenick PA, Boggs DR, Marsh JC, Cartwright GE, Wintrobe MM. Quantitative studies of blood and bone marrow neutrophils in normal mice. Am J Physiol. 1968;215: 353-360. [DOI] [PubMed] [Google Scholar]
- 31.Mitruka BM, Rawnsley HM. Clinical, Biochemical and Hematological Reference Values in Normal Experimental Animals and Normal Humans. New York, NY: Masson; 1981.
- 32.Semerad CL, Liu F, Gregory AD, Stumpf K, Link DC. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity. 2002;17: 413-423. [DOI] [PubMed] [Google Scholar]
- 33.Wooten DK, Xie X, Bartos D, Busche RA, Longmore GD, Watowich SS. Cytokine signaling through Stat3 activates integrins, promotes adhesion, and induces growth arrest in the myeloid cell line 32D. J Biol Chem. 2000;275: 26566-26575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim CH, Broxmeyer HE. In vitro behavior of hematopoietic progenitor cells under the influence of chemoattractants: stromal cell-derived factor-1, steel factor, and the bone marrow environment. Blood. 1998;91: 100-110. [PubMed] [Google Scholar]
- 35.Hestdal K, Ruscetti FW, Ihle JN, et al. Characterization and regulation of RB6–8C5 antigen expression on murine bone marrow cells. J Immunol. 1991;147: 22-28. [PubMed] [Google Scholar]
- 36.Tamura M, Hattori K, Nomura H, Oheda M, Kubota N. Induction of neutrophilic granulocytosis in mice by administration of purified human native granulocyte colony-stimulating factor (G-CSF). Biochem Biophys Res Commun. 1987;142: 454-460. [DOI] [PubMed] [Google Scholar]
- 37.Roberts AW, Metcalf D. Granulocyte colony-stimulating factor induces selective elevations of progenitor cells in the peripheral blood of mice. Exp Hematol. 1994;22: 1156-1163. [PubMed] [Google Scholar]
- 38.Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI. Chemokine binding and activities mediated by the mouse IL-8 receptor. J Immunol. 1995;155: 2158-2164. [PubMed] [Google Scholar]
- 39.Bozic CR, Gerard NP, von Uexkull-Guldenband C, et al. The murine interleukin 8 type B receptor homologue and its ligands: expression and biological characterization. J Biol Chem. 1994;269: 29355-29358. [PubMed] [Google Scholar]
- 40.Boulay R, Tardif M, Brouchon L, Vignais P. The human N-formylpeptide receptor: characterization of two cDNA isolates and evidence for a new subfamily of G-protein-coupled receptors. Biochemistry. 1990;29: 11123-11133. [DOI] [PubMed] [Google Scholar]
- 41.Gao J-L, Lee EJ, Murphy PM. Impaired anitbacterial host defense in mice lacking the N-formylpeptide receptor. J Exp Med. 1999;189: 657-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiner OD, Servant G, Welch MD, Mitchison TJ, Sedat JW, Bourne HR. Spatial control of actin polymerization during neutrophil chemotaxis. Nat Cell Biol. 1999;1: 75-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin Ther Targets. 2004;8: 409-422. [DOI] [PubMed] [Google Scholar]
- 44.Burdon PC, Martin C, Rankin SM. The CXC chemokine MIP-2 stimulates neutrophil mobilization from the rat bone marrow in a CD49d-dependent manner. Blood. 2005;105: 2543-2548. [DOI] [PubMed] [Google Scholar]
- 45.Hou SX, Zheng Z, Chen X, Perrimon N. The JAK/STAT pathway in model organisms: emerging roles in cell movement. Dev Cell. 2002;3: 765-778. [DOI] [PubMed] [Google Scholar]
- 46.Silver DL, Naora H, Liu J, Cheng W, Montell DJ. Activated signal transducer and activator of transcription (STAT) 3: localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004;64: 3550-3558. [DOI] [PubMed] [Google Scholar]
- 47.Sano S, Itami S, Takeda K, et al. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999;18: 4657-4668. [DOI] [PMC free article] [PubMed] [Google Scholar]