

Abstract
Helicobacter pylori colonizes the stomach of half of the world's population, causing a wide spectrum of disease ranging from asymptomatic gastritis to ulcers to gastric cancer. Although the basis for these diverse clinical outcomes is not understood, more severe disease is associated with strains harboring a pathogenicity island. To characterize the genetic diversity of more and less virulent strains, we examined the genomic content of 15 H. pylori clinical isolates by using a whole genome H. pylori DNA microarray. We found that a full 22% of H. pylori genes are dispensable in one or more strains, thus defining a minimal functional core of 1281 H. pylori genes. While the core genes encode most metabolic and cellular processes, the strain-specific genes include genes unique to H. pylori, restriction modification genes, transposases, and genes encoding cell surface proteins, which may aid the bacteria under specific circumstances during their long-term infection of genetically diverse hosts. We observed distinct patterns of the strain-specific gene distribution along the chromosome, which may result from different mechanisms of gene acquisition and loss. Among the strain-specific genes, we have found a class of candidate virulence genes identified by their coinheritance with the pathogenicity island.
Helicobacter pylori is a highly host-adapted bacterial pathogen that establishes a chronic infection in the human stomach and has no known animal or environmental reservoirs (1). Epidemiological and serological studies have revealed that H. pylori strains containing the CagA protein are associated with more severe disease (2) and harbor a 40-kb pathogenicity island (PAI) (3, 4). The PAI encodes a bacterial type IV secretory system that secretes and translocates the CagA protein into host cells (5–8), where it is phosphorylated by a host-cell kinase and causes morphological changes (7). The PAI also induces IL-8 production by host cells independent of the CagA protein (9–11). Efforts to classify H. pylori strains further by DNA fingerprinting uncovered extensive diversity (12, 13). The sequencing of two H. pylori genomes from independent strains, both containing the PAI, revealed that much of this diversity is silent at the amino acid level and thus at the functional gene level (14, 15). Here we used a H. pylori DNA microarray to examine the genomic composition of H. pylori clinical isolates containing and lacking the PAI at the level of individual genes to characterize the extent of genetic diversity between strains and to search for new candidate virulence determinants.
Materials and Methods
PCR Primer Design.
The elements of our microarray consisted of large (mean size, 817 base pairs; 10th percentile, 130 base pairs; 90th percentile, 1,967 base pairs) DNA fragments corresponding to unique segments of individual open reading frames (ORFs). These fragments were generated by PCRs using gene-specific primers. We aimed to include in our array the superset of ORFs from both published genomes. When an ORF was present in both J99 and 26695, we arbitrarily chose 26695 as the reference strain, designing PCR primers from its published sequence and using its genomic DNA as a template in the amplifications. Strain J99 provided the sequence and template DNA for the PCRs of the 91 ORFs present only in that strain.
PCR primers were designed by using a modified version of the program web primer (a kind gift of Paul Spellman, Charles Scafe, and David Botstein, Stanford University). This program takes a list of ORF coordinates provided by the user and generates primer pairs for each satisfying user-determined parameter (maximum cross- and self-annealing scores, optimum melting temperatures, minimum distance from start and stop sites). To ensure that the elements of our array would detect specifically their corresponding genes and no others, the ORF coordinates fed into the primer program were circumscribed such that they would exclude regions of any ORF with high crosshomology to other regions of the genome. Such regions were identified by using blast_summary.pl, a program that parses the results of a gapped National Center for Biotechnology Information blast search into a tab-delimited file and generates a homology score, M, for each match:
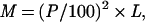 |
where P is the percent identity, and L is the length (in base pairs) of the match. For each ORF, the coordinates defining the largest contiguous region not containing a sequence with an M score greater than 50 (roughly equivalent to 50 base pairs of 100% identity) and not overlapping an adjacent ORF were used as inputs for the primer program. We were unable to define unique regions for the 16 different transposase genes, so these were each spotted onto the array. These transposase spots would be expected to cross-hybridize with other transposase gene sequences.
PCR.
Primer pairs were grouped by similar melting temperatures and synthesized in 96-well plates (SigmaGenosys, The Woodlands, TX). To improve amplification yield, universal 13-bp sequences were incorporated into the 5′ ends of each primer. Each upstream primer (gene relative) contained the sequence CCTGGTCGACTGC, and each downstream primer, CCACCTCGAGCAC. PCRs were performed in two rounds, the second being a repeat of reactions that failed in the first, under less stringent magnesium and temperature conditions. In the first round, reactions (70 μl) consisted of the following: 5–7 ng of genomic DNA, 30 mM Tricine (pH 8.5), 1.5 mM magnesium chloride, 50 mM potassium chloride, 0.25 mM each dNTP, and 20 pmol each primer. Round 1 reactions were carried out in an MJ Research (Cambridge, MA) PTC-225 DNA Engine Tetrad thermal cycler under the following conditions: 94°C 2 min, followed by 30 rounds of 30 seconds at 94°C, 30 seconds at the annealing temperature, and 4 min at 72°C, followed by one round of 10 min at 72°C. Annealing temperatures were equal to the lowest melting temperature for the primers in a given plate (range: 50–56°C). For round 2, the annealing temperatures were reduced to 50°C for all primer pairs and magnesium concentrations increased to 2.5 mM. Amplification success was checked visually after agarose gel electrophorisis for intensity and correct size. The success rate was 85.2% after these rounds. At this point, primers that failed to yield products were redesigned, and two more rounds of amplification were attempted. After this redesign, 98.9% of all ORFs (1,660/1,681) were successfully amplified.
Array Printing.
All steps of PCR product precipitation, resuspension and dilution, glass slide preparation, printing, and processing after printing were performed as described (16). The final array contained 1,660 unique sequences printed twice.
Bacterial Strains and Growth.
H. pylori were obtained from frozen stocks and grown on solid media on horse-blood plates containing 4% Columbia agar base (Oxoid, Hampshire, U.K.), 5% defibrinated horse blood (HemoStat Labs, Dixon, CA), 0.2% β-cyclodextrin (Sigma), 10 μg/ml vancomycin (Sigma), 5 μg/ml cefsulodin (Sigma), 2.5 units/ml polymyxin B (Sigma), 50 μg/ml cyclohexamide (Sigma), 5 μg/ml trimethoprim (Sigma), and 8 μg/ml amphotericin B (Sigma) under microaerobic conditions at 37°C. A microaerobic atmosphere was generated either by using a CampyGen sachet (Oxoid) in a gas pack jar or by incubation in an incubator equilibrated with 10% CO2 and 90% air. All strains were obtained from the sources indicated except strains AR32 and H34, which were clinical isolates from patients undergoing endoscopy at Stanford Hospital obtained from J. Parsonnet, Stanford University.
Preparation and Hybridization of Genomic DNA Probes.
Two micrograms of genomic DNA (for test strains), prepared from plate-grown bacteria by a Wizard genomic DNA purification kit (Promega) or 1 μg each of 26695 and J99 genomic DNA prepared by CsCl gradient centrifugation (reference sample), were suspended in a total volume 38 μl H2O and denatured 5 min 99°C. Five microliters of 10× Buffer (400 μg/ml random octamers/0.5 M Tris⋅HCL/100 mM MgSO4/10 mM DTT)/5 μl dNTP/dUTP mix (0.5 mM dGTP, dATP, dCTP, 0.2 mM aminoallyl dUTP/0.3 mM dTTP)/2 μl Klenow was added and the reaction incubated 1 h at 37°C. Free amines were removed by adding the reaction (50 μl) to a Microconym YM 30 (Millipore) containing 450 μl of H2O. The samples were concentrated by centrifuging for 8 min at 11,750 × g in a microcentrifuge. The eluate was discarded and the wash step repeated two times. After the last wash, the samples were collected and dried in a Speed Vac (Savant). The probe was resuspended in 4.5 μl H2O and labeled by the addition of 4.5 μl of 0.1 M sodium bicarbonate, pH 9.0, containing 1/16 of one reaction vial FluoroLink Cy5 or Cy3 monofunctional dye (Amersham), and was incubated for 1 h at room temperature in the dark. The reaction was quenched by addition of 4.5 μl of 4 M hydroxylamine and incubated for 15 min at room temperature in the dark. The Cy3 and Cy5 reactions were combined and unincorporated dye removed by using a Qia-Quick PCR purification column according to the manufacturer's instructions (Qiagen, Chatsworth, CA). The eluate from the columns was dried in a Speed Vac and resuspended in 11 μl TE (10 mM Tris, pH 8.0/1 mM EDTA). To this was added 1 μl of 25 mg/ml yeast tRNA, 2.55 μl of 20× SSC (3 M NaCl/0.3 M trisodium citrate 2 H2O, pH 7), and 0.45 μl of 10% SDS. The mixture was denatured 2 min at 99°C, cooled briefly, and centrifuged for 2 min at 13,800 × g in a microcentrifuge. The probes were applied to the microarrays for hybridization and washed as described (16). The microarrays were scanned by using an Axon scanner with genepix 3.0 software (Axon Instruments, Redwood City, CA) and further processed by using scanalyze 2.44 (http://www.microarrays.org/software.html; ref. 17) or genepix 3.0 software. Data for each channel of the microarray were normalized by using the default-computed normalization of the Stanford Microarray Database (http://genome-www4.stanford.edu/MicroArray/help/results_normalization.html).
Determination of Gene Content of Strains and Data Analysis.
For each test strain, we computed the geometric mean of the normalized red/green (R/G) ratio by using data from two to three arrays yielding two to six readings per gene (each array contains two spots for each gene). Spots were excluded because of slide abnormalities or low signal (<100 arbitrary units in the green channel). We chose a cutoff for the normalized R/G ratio <0.5 for the absence of a gene based on test hybridizations of strain 26695 and J99 vs. the reference sample (an equimolar ratio of 26695 and J99 DNA). We predicted the hybridization results on the basis of our modified annotation of the two sequenced strains where we designated genes common to both strains if they had greater than 85% identity to the other genome over 60% of their length (14 sequences from J99 and 53 sequences from 26695 were reassigned from strain specific to common). With this cutoff, we had 0% false positives (0/73) and 1% false negatives (15/1,570) for 26695 and 7% false positives (7/105) and 2% false negatives (32/1,570) for J99. As expected, strain 26695 gave a lower error rate because its sequence was used to design the majority of the spots on the array. Genes with readings in fewer than 12 strains (80%) were excluded from the analysis, leaving 1,643 genes in our data set. The data were simplified into a binary score (gene present in a given strains = 1, absent = 0) and analyzed by average hierarchical clustering by using the cluster program and displayed by using the treeview program (http://www.microarrays.org/software.html; ref. 17). The complete data set used is available (see supplemental data at www.pnas.org). To assign the homology classes of the genes, blast searches of the nonredundant database (GenBank) were performed for the amino acid sequences of all ORFs on the array. Previous annotations were used for genes of known function (14, 15). Those with unknown function were termed “conserved” if they had a match in the database with a P value <10−10. Covariance was calculated by computing the absolute difference between the binary score for gene M vs. gene N for strain S (Δ score). The covariance index for gene M vs. gene N was calculated as 1 − [sum(Δ score for strains 1–15)/number of strains assayed].
Results and Discussion
Comparison of the two H. pylori genome sequences reveals that, whereas most of the genes are highly conserved between the two strains (26695 and J99), approximately 6% of the genes in each genome are not found in the other (14, 18, 19). We constructed an H. pylori DNA microarray containing 98% of the ORFs of both sequenced strains. Care was taken in PCR primer design to avoid amplifying regions in a given gene with cross-homology to other genes in the genome. This allowed us, for example, to distinguish the individual genes encoding members of a highly conserved outer membrane protein family by their unique 3′ sequences. The final array contained 1,660 unique sequences printed twice. To test the discriminatory power of our arrays, we hybridized them with genomic DNA from 26695 and J99. We could detect the presence of the strain-specific genes with 98% specificity and 96% sensitivity.
We examined the extent of genetic diversity among H. pylori strains by analyzing the genome composition of 15 strains (Table 1). Our experimental design for whole-genome scanning was to perform comparative hybridizations between a Cy3-labeled reference mixture of genomic DNA from the two sequenced strains—J99 and 26695—and Cy5-labeled genomic DNA of the strain being tested. Of the 1,643 genes analyzed, 1,281 were common to all strains, representing the functional core of the proteome. In contrast, 362 (22%) were missing from at least one strain (strain-specific genes), including 184 genes previously identified in both sequenced strains, thus doubling the number of H. pylori strain-specific genes. These strain-specific genes may represent those with redundant functions required for specific niches or whose DNA sequences vary so much between strains they cannot be detected under our hybridization conditions. To estimate how well our data set approximated the number of genes in the functional core, we randomly generated 50 independent groups of strains of size 1–15, and the average number of strain-specific genes was calculated as a function of the number of strains. The rate of increase of strain-specific genes could be approximated as an exponentially decreasing function that fell below the level of the number of false negatives at seven strains. In addition to the core 1,281 genes, each strain contained 168–275 strain-specific genes, thus at least 12–18% of each strain's genome was composed of strain-specific genes (Table 1). Strains likely contain other strain-specific genes not present in the 26695 and J99 sequences and thus not represented on the array.
Table 1.
Strains
| Strain | Origin | Total no. of genes present from microarray | No. of J99-specific genes | No. of 26695-specific genes | Ref. |
|---|---|---|---|---|---|
| 26695 | U.K., gastritis | 1,556 | 0 | 105 | 15 |
| J99 | U.S., ulcer | 1,511 | 73 | 7* | 14 |
| 87A300 | U.S. | 1,487 | 26 | 44 | 29 |
| AR32 | U.S., ulcer | 1,510 | 48 | 36 | This study |
| H34 | U.S., gastritis | 1,449 | 31 | 37 | This study |
| HP1 | Peru, ulcer | 1,506 | 43 | 52 | 30 |
| SPM-292 | Italy | 1,453 | 19 | 44 | 31 |
| SPM-314 | Italy | 1,526 | 58 | 57 | 31 |
| SPM-326 | Italy | 1,514 | 24 | 61 | 31 |
| SPM-342 | Italy | 1,477 | 13 | 55 | 31 |
| G27 | Italy | 1,476 | 24 | 53 | 32 |
| G39 | Italy | 1,488 | 25 | 44 | 32 |
| G50 | Italy | 1,499 | 52 | 54 | 32 |
| SS1 | Australia | 1,463 | 28 | 40 | 33 |
| NCTC11638 | Australia | 1,502 | 23 | 57 | 34 |
| H. pylori microarray | 1,643 | 73 | 105 |
These represent false-positive measurements as described in Materials and Methods.
The H. pylori functional core contained most of the genes encoding metabolic, biosynthetic, cellular, and regulatory functions (Fig. 1). The most abundant class of strain-specific genes was unique to H. pylori with no known function and were enriched among the strain-specific genes (58%) vs. the core genes (21%). In contrast, genes of unknown function but with homology to other bacterial genes were enriched in the functional core (16%) vs. the strain-specific genes (7%). The homologies of other classes of strain-specific genes implied functions that may aid the bacterium under certain circumstances or in certain hosts. For example, one strain-specific gene was the babA outer-membrane protein (OMP), which encodes an adhesion for the Lewis B antigen present on gastric tissue of some individuals (20). Eight additional OMP genes and seven lipopolysaccharide synthesis genes were among the strain-specific genes. The two largest classes of strain-specific genes with known function (restriction modification system components and transposase genes) encoded genes that regulate DNA exchange among bacteria and may promote the genetic diversification of H. pylori (21, 22). The remaining strain-specific genes include three virB4 homologues and a traG homologue, both ATPases involved in assembly of type IV secretion structures, and three topoisomerases.
Figure 1.

Homology classes of H. pylori core and strain-specific genes. Each bar represents the number of strain-specific (gray) and core genes (white) in each of the following homology classes (number of genes/percentage of genes): unknown, H. pylori specific strain-specific (210/56%) core (267/21%); metabolism and biosynthesis, excluding DNA metabolism, strain-specific (11/3%) core (410/32%) strain specific; unknown, conserved strain-specific (27/7%) core (205/16%); cell envelope, excluding OMP and lipopolysaccharide (LPS), strain-specific (0/0%) core (157/12%); DNA metabolism, strain-specific (8/2%) core (54/4%); cellular process, strain-specific (1/0.3%) core (55/4%); restriction modification, strain-specific (38/10%) core (14/1%); OMP, strain-specific (9/2%) core (42/3%); regulatory function, strain-specific (0/0%) core (38/3%); LPS biosynthesis, strain-specific (7/2%) core (29/2%); PAI, unknown function, strain-specific (20/6%) core (0/0%); type IV secretion and conjugation, strain-specific (12/3%) core (6/0.5%); transposase, strain-specific (16/4%) core (0/0%); other, strain-specific (3/0.8%) core (4/0.3%).
Because gene order is largely conserved between the two sequenced strains (14), we examined the pattern of strain-specific genes ordered along an H. pylori chromosome map (Fig. 2a). Many of the strain-specific genes were found in two large regions, the PAI and the plasticity zone (PZ), which were previously noted because of their different G + C content, a hallmark of horizontally acquired sequences (14, 15, 23). Our analysis suggested that the PAI is largely inherited as a single block of 27 genes, although one strain contained only 2 PAI genes and another, 7 PAI genes. In contrast, the PZ appears highly mosaic and contains multiple transposases and endonucleases, suggesting that it is a site of extensive insertion, excision, and recombination. Whereas the majority of the 26695 and J99 strain-specific genes were located in the PZ, we found two-thirds of all of the strain-specific genes distributed elsewhere in the chromosome in smaller tracts of 1 to 8 genes. The homologies of the genes found in these smaller tracts correlated with the size of the locus (Table 2). For example, whereas most of the OMPs and lipopolysaccharide biosynthesis strain-specific genes were flanked by conserved genes, the restriction modification genes, some of which have been postulated to behave as selfish operons (24), were found most often in cassettes of two to seven genes. All of the transposases were found within larger tracts of strain-specific genes (more than three), suggesting they ferry additional genes when they insert into a genome.
Figure 2.

Chromosomal position and composition of strain-specific genes. The genes were ordered by using a combined J99/26695 map where J99-specific genes are placed at the appropriate sites in the 26695 chromosomal map. The presence (blue) or absence (yellow) of genes was displayed according to their position on the chromosome for each strain (missing data are gray). (Left) The entire H. pylori chromosome starting at position 1. (Right) Zoomed images of the two portions of the plasticity zone, which is continuous in the J99 genome, and the PAI. The names of genes with homology to transposases (orange), topoisomerases or endonucleases (turquoise), type IV secretion homologues (purple), or restriction modification systems (dark green) are indicated.
Table 2.
Distribution of strain-specific genes of different homology classes by locus size
| No. of genes per chromosomal locus* | Number of genes
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Total | Unknown, H. pylori specific | Unknown, Conserved | Other | Transposase | Restriction modification | LPS | OMP | |
| 1 | 68 | 41 | 4 | 8 | 0 | 4 | 3 | 8 |
| 2 | 56 | 33 | 5 | 4 | 0 | 11 | 2 | 1 |
| 3 | 36 | 21 | 2 | 3 | 2 | 8 | 0 | 0 |
| 4 | 16 | 11 | 0 | 1 | 0 | 2 | 2 | 0 |
| 5 | 15 | 9 | 0 | 2 | 3 | 1 | 0 | 0 |
| 6 | 30 | 19 | 0 | 1 | 4 | 6 | 0 | 0 |
| 7 | 14 | 7 | 0 | 0 | 2 | 5 | 0 | 0 |
| 8 | 8 | 8 | 0 | 0 | 0 | 0 | 0 | 0 |
| PAI (27) | 27 | 0 | 0 | 27 | 0 | 0 | 0 | 0 |
| PZ1 (45) | 45 | 34 | 2 | 6 | 2 | 1 | 0 | 0 |
| PZ2 (57) | 57 | 41 | 0 | 6 | 9 | 1 | 0 | 0 |
LPS, lipopolysaccharide.
The locus size refers to the number of adjacent strain-specific genes. Not all genes in a given locus may be missing in any given strain.
Evolutionary pressures select for the coinheritance of genes involved in common pathways, thus identification of covarying genes can reveal functional relationships not evident from sequence alone (25). We therefore used a hierarchical clustering program (17) to generate a dendogram of genes based on their patterns of absence and presence across the strains to detect such functional groups. Although the majority of gene clusters lacked informative homologies, the 27 PAI genes, which together secrete and deliver at least one bacterial protein to host cells, clustered together with a Pearson correlation coefficient >0.75 by this analysis (Fig. 3). Interestingly, seven additional genes located elsewhere in the chromosome clustered with the PAI genes, including two OMPS: omp27 and babA. We independently calculated a covariance index (CI) for all of the strain-specific genes with each other across the 15 strains (see Materials and Methods). Averaging the CI values for the PAI genes identified 10 genes that covary with the PAI genes (CIave > 0.8) (Table 3), including the 7 genes that cocluster in Fig. 3. The BabA adhesin is a known virulence factor previously shown to covary with the PAI (20), and it is correlated with disease severity (26). The other nine genes represent new potential virulence factors that may modulate PAI functions, such as induction of IL-8 production by host cells, or act synergistically with the PAI to cause bacterial persistence or disease. Four genes had low values (CIave < 0.2; Table 3), indicating they are infrequently present with the PAI. Two of these genes have homologies to restriction modification genes and may function directly or indirectly to prevent acquisition of the PAI.
Figure 3.

Cluster analysis of PAI genes. The subset of 362 strain-specific genes was used to generate a dendogram of both the strains and the genes based on the presence or absence of genes. The strains and genes were grouped by average hierarchical clustering using the cluster program and the output displayed using the treeview program. The portion of the cluster containing the PAI genes is shown where the presence (blue) or absence (yellow) of genes is indicated. Genes within the PAI (purple) and genes with homology to OMPs (blue), restriction modification genes (green), or no homology (black) are indicated. (Left) Dendogram of genes that cocluster with the PAI genes, with the Pearson correlation coefficients. Above the cluster is the dendogram of H. pylori strains, based on all strain-specific genes, with the Pearson correlation coefficients. The entire cluster is available as Table 4, which is published as supplemental data on the PNAS web site www.pnas.org.
Table 3.
Average CIave of genes that covary or antivary with PAI genes
| Gene identification | Gene name | Gene description | CIave |
|---|---|---|---|
| CIave > 0.8 | |||
| HP0528–46 | cag8–25 | PAI genes | 0.973 |
| HP0522–27 | cag3,4,7, virD4, virB11 | PAI genes | 0.946 |
| HP0547 | cag26 (cagA) | PAI gene | 0.916 |
| HP0965 | 0.906 | ||
| HP0483 | Type II DNA methyltransferase | 0.889 | |
| HP0484 | 0.889 | ||
| HP0520 | cag1 | PAI gene | 0.889 |
| HP1177 | omp27 | OMP | 0.889 |
| HP0521 | orf7 | PAI gene | 0.884 |
| HP1243 | omp28 (babA) | OMP, adhesin | 0.884 |
| HP0260 | mod | Type II DNA methyltransferase | 0.879 |
| HP0186 | 0.822 | ||
| HP0767 | 0.822 | ||
| HP0963 | 0.813 | ||
| HP1417 | 0.813 | ||
| CIave < 0.2 | |||
|---|---|---|---|
| JHP0046 | Putative Type II restriction enzyme | 0.188 | |
| JHP0045 | Putative Type II DNA methyltransferase | 0.188 | |
| HP1426 | Conserved hypothetical protein | 0.183 | |
| HP0670 | 0.160 |
CI, covariance index.
We also used hierarchical clustering to explore the relationship of the different strains based on their gene complement (Fig. 3). Although the strains fell into distinct groups in the dendogram, the small sample size and lack of clinical information for many of the strains did not allow us to correlate these groups with particular disease outcomes or geographic origin. Previous H. pylori strain typing has been based on the presence of the CagA gene in the PAI. We found that PAI-lacking strains were not more related to each other than to PAI-containing strains, suggesting that the PAI does not define an evolutionary lineage. This finding, as well as the lack of mosaicism in the PAI (Fig. 2), is consistent with observations of the loss of the PAI in a subpopulation of bacteria isolated from a single infected individual (27).
H. pylori can reside for decades in the exclusive niche of the human stomach with few competing resident bacteria, where it can diversify as a population, mutate, excise sequences, and sample the genetic material of transient superinfecting H. pylori strains and other bacteria (12, 28). H. pylori has a small genome (1.7 Mb) with a paucity of transcriptional regulators. Our analysis reveals a conserved core of 1,281 genes and an additional 362 strain-specific genes that compose up to 18% of a given strain's genome. Although our experimental system was able to approximate the number of core genes, it certainly underestimated the number of strain-specific genes, as our microarray contains only genes present in at least one of the sequenced strains. The strain-specific genes may encode adaptations to genetically diverse hosts or factors contributing to different disease outcomes. Examination of their distribution along the chromosome indicated that H. pylori uses multiple mechanisms of gene acquisition and loss that may contribute to its phenotypic diversity. Finally, our analysis of strain-specific gene covariance allowed us to identify candidate virulence genes coinherited with the PAI that can now be tested for modulation of PAI activity in cellular assays and for their role during infection of animal models. Extension of this analysis to larger strain collections from case-controlled studies should uncover further nuances of H. pylori's genetic program that are important for specific disease outcomes.
Supplementary Material
Acknowledgments
We thank Simon Sims of SigmaGenosys for primer synthesis, Paul Spellman and Charles Scafe for providing primer design software, Ian Manger for advice and help in array printing and database development, David Botstein and Arkady Khodursky for strategic advice, Charles Kim for help in programming, Eric Johnson and Phil Beineke for help with data analysis, Inna Billis for technical assistance, Jeffrey Gordon (Washington University, St. Louis, MO), Antonello Covacci (Chiron-Biocine, Siena, Italy), Julie Parsonnet (Stanford University), and Adrian Lee (University of New South Wales, Sydney, Australia) for strains and helpful discussions, and Lalita Ramakrishnan for critical review of the manuscript. We acknowledge support from the Jane Coffin Childs memorial fund for medical research to N.S., from the Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation (Fellowship DRG-1456 to T.M. and DRG-1509 to K.G.), National Institutes of Health Grant AI38459 to L.T., and contract with Protein Design Labs to S.F.
Abbreviations
- ORF
open reading frame
- PAI
pathogenicity island
- OMP
outer-membrane protein
References
- 1.Covacci A, Telford J L, Del Giudice G, Parsonnet J, Rappuoli R. Science. 1999;284:1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- 2.Xiang Z, Censini S, Bayeli P F, Telford J L, Figura N, Rappuoli R, Covacci A. Infect Immun. 1995;63:94–98. doi: 10.1128/iai.63.1.94-98.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akopyants N S, Clifton S W, Kersulyte D, Crabtree J, Youree B E, Reece C A, Bukanov N O, Drazek E S, Roe B A, Berg D E. Mol Microbiol. 1998;28:37–53. doi: 10.1046/j.1365-2958.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- 4.Censini S, Lange C, Xiang Z, Crabtree J E, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. Proc Natl Acad Sci USA. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, Tsubokawa M, Tohyama Y, Maeda S, Omata M, et al. J Exp Med. 2000;191:593–602. doi: 10.1084/jem.191.4.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stein M, Rappuoli R, Covacci A. Proc Natl Acad Sci USA. 2000;97:1263–1268. doi: 10.1073/pnas.97.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segal E D, Cha J, Lo J, Falkow S, Tompkins L S. Proc Natl Acad Sci USA. 1999;96:14559–14564. doi: 10.1073/pnas.96.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 9.Glocker E, Lange C, Covacci A, Bereswill S, Kist M, Pahl H L. Infect Immun. 1998;66:2346–2348. doi: 10.1128/iai.66.5.2346-2348.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crabtree J E, Covacci A, Farmery S M, Xiang Z, Tompkins D S, Perry S, Lindley I J D. J Clin Pathol. 1995;48:41–45. doi: 10.1136/jcp.48.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crowe S E, Alvarez L, Dytoc M, Hunt R H, Muller M, Sherman P, Patel J, Jin Y, Ernst P B. Gastroenterology. 1995;108:65–74. doi: 10.1016/0016-5085(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 12.Marshall D G, Dundon W G, Beesley S M, Smyth C J. Microbiology. 1998;144:2925–2939. doi: 10.1099/00221287-144-11-2925. [DOI] [PubMed] [Google Scholar]
- 13.Alm R A, Trust T J. J Mol Med. 1999;77:834–846. doi: 10.1007/s001099900067. [DOI] [PubMed] [Google Scholar]
- 14.Alm R A, Ling L-S L, Moir D T, King B L, Brown E D, Doig P C, Smith D R, Noonan B, Guild B C, Dejonge B L, et al. Nature (London) 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 15.Tomb J-F, White O, Kerlavage A R, Clayton R A, Sutton G G, Fleischmann R D, Ketchum K A, Klenk H P, Gill S, Dougherty B A, et al. Nature (London) 1997;338:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 16.Eisen M B, Brown P O. Methods Enzymol. 1999;303:179–205. doi: 10.1016/s0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- 17.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doig P, de Jonge B L, Alm R A, Brown E D, Uria-Nickelsen M, Noonan B, Mills S D, Tummino P, Carmel G, Guild B C, et al. Microbiol Mol Biol Rev. 1999;63:675–707. doi: 10.1128/mmbr.63.3.675-707.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marais A, Mendz G L, Hazell S L, Megraud F. Microbiol Mol Biol Rev. 1999;63:642–674. doi: 10.1128/mmbr.63.3.642-674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ilver D, Arnqvist A, Ogren J, Frick I M, Kersulyte D, Incecik E T, Berg D E, Covacci A, Engstrand L, Boren T. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 21.Xu Q, Morgan R D, Roberts R J, Blaser M J. Proc Natl Acad Sci USA. 2000;97:9671–9676. doi: 10.1073/pnas.97.17.9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akopyants N S, Fradkov A, Diatchenko L, Hill J E, Siebert P D, Lukyanov S A, Sverdlov E D, Berg D E. Proc Natl Acad Sci USA. 1998;95:13108–13113. doi: 10.1073/pnas.95.22.13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ochman H, Lawrence J G, Groisman E A. Nature (London) 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi I. Trends Genet. 1998;14:368–374. doi: 10.1016/s0168-9525(98)01532-7. [DOI] [PubMed] [Google Scholar]
- 25.Pellegrini M, Marcotte E M, Thompson M J, Eisenberg D, Yeates T O. Proc Natl Acad Sci USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Proc Natl Acad Sci USA. 1999;96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Figura N, Vindigni C, Covacci A, Presenti L, Burroni D, Vernillo R, Banducci T, Roviello F, Marrelli D, Biscontri M, et al. Gut. 1998;42:772–778. doi: 10.1136/gut.42.6.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuipers E J, Israel D A, Kusters J G, Gerrits M M, Weel J, van Der Ende A, van Der Hulst R W, Wirth H P, Hook-Nikanne J, Thompson S A, Blaser M J. J Infect Dis. 2000;181:273–282. doi: 10.1086/315173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Segal E D, Shon J, Tompkins L S. Infect Immun. 1992;60:1883–1889. doi: 10.1128/iai.60.5.1883-1889.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guruge J L, Falk P G, Lorenz R G, Dans M, Wirth H-P, Blaser M J, Berg D E, Gordon J I. Proc Natl Acad Sci USA. 1998;95:3925–3930. doi: 10.1073/pnas.95.7.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchetti M, Arico B, Burroni D, Figura N, Rappuoli R, Ghiara P. Science. 1995;267:1655–1658. doi: 10.1126/science.7886456. [DOI] [PubMed] [Google Scholar]
- 32.Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N, Rappuoli R. Proc Natl Acad Sci USA. 1993;90:5791–5795. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee A, O'Rourke J, De Ungria M C, Robertson B, Daskalopoulos G, Dixon M F. Gastroenterology. 1997;112:1386–1397. doi: 10.1016/s0016-5085(97)70155-0. [DOI] [PubMed] [Google Scholar]
- 34.Akopyants N S, Jiang Q, Taylor D E, Berg D E. Helicobacter. 1997;2:48–52. doi: 10.1111/j.1523-5378.1997.tb00058.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.