

Abstract
A method for sensitively monitoring enzyme kinetics and activities by using dual-color fluorescence cross-correlation spectroscopy is described. This universal method enables the development of highly sensitive and precise assays for real-time kinetic analyses of any catalyzed cleavage or addition reaction, where a chemical linkage is formed or cleaved through an enzyme’s action between two fluorophores that can be discriminated spectrally. In this work, a homogeneous assay with restriction endonuclease EcoRI and a 66-bp double-stranded DNA containing the GAATTC recognition site and fluorophores at each 5′ end is described. The enzyme activity can be quantified down to the low picomolar range (>1.6 pM) where the rate constants are linearly dependent on the enzyme concentrations over two orders of magnitude. Furthermore, the reactions were monitored on-line at various initial substrate concentrations in the nanomolar range, and the reaction rates were clearly represented by the Michaelis–Menten equation with a KM of 14 ± 1 nM and a kcat of 4.6 ± 0.2 min−1. In addition to kinetic studies and activity determinations, it is proposed that enzyme assays based on the dual-color fluorescence cross-correlation spectroscopy will be very useful for high-throughput screening and evolutionary biotechnology.
Kinetic studies on enzymes are among the most important tools for understanding biological interactions at the molecular level. In combination with new approaches in genetic engineering and structure determination, there have been major efforts in recent years to develop more sensitive and precise techniques for characterizing the kinetics of enzyme reactions. These techniques have made it possible to develop efficient assays for analyzing catalytic parameters such as turnover rates, substrate specificity, as well as regio- and stereospecificity; they constitute a major part of the biochemical and pharmaceutical research. Moreover, the alteration of catalytic properties, either by evolutionary biotechnology (1–4) or by rational approaches, is of special interest for medical and industrial applications. The desired features are as follows: accelerated reaction rates; higher, relaxed, or even new substrate specificities; tolerance to nonnatural environments; and novel types of reactions. Modern rational design is the interplay between computer modeling and experimental testing of designed molecules by sensitive and quantitative assays. On the other hand, evolutionary approaches make use of rapid and highly sensitive screening assays to detect minute quantities of better adapted individuals among a large excess of alternatives. For all these purposes, fluorescence correlation spectroscopy (FCS) (5–8), with its ability to quantify molecular interactions at nanomolar and lower concentrations, has been proposed as an ideal tool (2). Most contemporary applications of FCS that are performed in confocal microscope (9) are based on the analysis of the molecular dynamics and the reaction kinetics of fluorescently labeled biomolecules that undergo temporal changes in their diffusion properties (10–17). The dual-color cross-correlation method, as proposed by Rigler and Eigen (2, 18), circumvents the necessity of evaluating the diffusion characteristics of the product fractions; therefore, simple mathematical evaluation (18, 19) and much shorter readout times for screening applications can be achieved as shown in the subsequent article (20). The instrumentation setup allows one to sensitively detect molecules that bear two different fluorescent labels. This can be achieved by a proper combination of excitation and detection schemes utilizing two fluorescent labels that are spectrally well separated. The experiment is designed in such a way that the reaction step to be investigated converts doubly labeled educts in singly labeled products or vice versa. In this study we used the cleavage of a double-stranded DNA (dsDNA) molecule catalyzed by the restriction endonuclease EcoRI (21) to demonstrate the potential of dual-color fluorescence cross-correlation spectroscopy (dual-color FCS) for analyzing enzyme kinetics sensitively and reproducibly. The DNA molecule was labeled with a red and a green dye at opposite ends, and the catalyzed reaction was monitored on-line by using a dual-color fluorescence cross-correlation spectrometer. Due to enzymatic cleavage, the number of doubly labeled DNA substrate molecules contributing to the cross-correlation signal decreased continuously. The catalytic activity was detected accurately down to an enzyme concentration of about 1 pM. Digestion kinetics were properly described by the Michaelis–Menten equation, demonstrating the high suitability of this method.
MATERIALS AND METHODS
Materials.
Restriction endonuclease EcoRI (EC 3.1.21.4) was purchased from Stratagene, with an activity of 25 units/μl (one unit is defined as the amount required to completely digest 1.0 μg of λ DNA in 1 h at 37°C) in a storage buffer containing 300 mM NaCl, 5 mM Tris-acetate (pH 7.4), 0.1 mM EDTA, 5 mM β-mercaptoethanol, 0.15% Triton X-100, 200 μg/ml BSA, and 50% glycerol. Identity, purity, and concentration of the enzyme were proven by SDS/PAGE and HPLC (data not shown). The enzyme concentrations below are given in terms of the 62-kDa dimer. The two complementary 66-nt DNA oligonucleotides (Fig. 1) were custom-synthesized and HPLC purified by MWG-Biotech (Ebersberg, Germany), both unmodified and labeled at their 5′ ends with fluorescence dyes Cy5 (Amersham) and Rhodamine Green (Molecular Probes), respectively. Oligonucleotide concentrations were determined by UV absorption at 260 nm (ɛ = 8.0 × 105 M−1⋅cm−1), concentrations of Rhodamine Green at 510 nm (ɛ = 7.9 × 104 M−1⋅cm−1) and of Cy5 at 650 nm (ɛ = 2.5 × 105 M−1⋅cm−1). Complementary strands were annealed at 450 nM in a buffer containing 100 mM KOAc, 25 mM Tris-acetate (pH 7.6), 10 mM MgOAc, 0.5 mM β-mercaptoethanol, and 10 μg/ml BSA, by heating at 95°C for 2 min, gradually cooling down to 23°C with a slope of 1.2°C/min, and keeping this temperature for 10 min.
Figure 1.

Sequence of the dsDNA substrate with fluorophore labels. The EcoRI recognition sequence is shown in bold letters. Arrows indicate the phosphodiester bonds that are cleaved.
Endonucleolytic Assay and FCS Setup.
Reactions were carried out in a 40 μl volume in coverglass chambers (Nunc), by using a reaction buffer containing 150 mM KOAc, 37.5 mM Tris-acetate (pH 7.6), 15 mM MgOAc, 0.75 mM β-mercaptoethanol, 515 μg/ml BSA, 0.05% Triton X-100, 0.5% glycerol, and different amounts of DNA substrates. Enzyme solutions were diluted in the reaction buffer and added immediately before analysis. Reference samples contained all components except the enzyme. The on-line auto- and cross-correlation measurements of all reactions were made by using a cross-correlation FCS spectrometer of which a prototype was developed in our laboratory (19) and which was commercialized by C. Zeiss (Dual-color ConfoCor; Zeiss). Samples were excited in the focal measurement volume element illuminated by two laser beams at 488 nm and 633 nm with a total power of 38 kW/cm2 and 31 kW/cm2, respectively. The red and green fluorescence emission signals were detected separately by two avalanche diodes after passing through a confocally located 30 μm pinhole (Fig. 2). Red and green photocount signals were cross-correlated in 60-s cycles.
Figure 2.

The dual-color FCS setup. Two parallel laser beams of an argon-ion laser (488 nm, 0.1 mW) and a helium-neon laser (633 nm, 0.05 mW) pass through a water-immersion microscope objective (×40, 1.2 NA) in an epi-illumination arrangement resulting in two superimposed focal spots in the sample, forming a confocal volume element in the femtoliter range. The emitted fluorescence light is collected by the microscope objective, separated from the excitation light by a dichroic mirror, and focused onto a pinhole by a lens. The pinhole, with an adjustable diameter, is located in the image plane of this lens and can be adjusted in the x-y-z axes. Fluorescence emission is parallelized, separated by a dichroic mirror into a green and a red fraction and refocused on two avalanche photo diodes.
Theory of FCS Cross-Correlation Analysis.
Correlation analysis of fluorescence signals in diffusion systems is actually analysis of molecular number fluctuations, made possible by small confocal measurement volumes, low concentrations of fluorophores (nM), and ultrasensitive detection devices such as single-photon counting avalanche diodes (22). The general expression for fluctuation auto- and cross-correlation functions is given by:
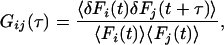 |
1 |
where 〈Fi(t)〉 and 〈Fj(t)〉 are the time-averaged fluorescence signals of two fluorescent species i and j; δFi and δFj denote their fluctuations at times t and t + τ. The single-color auto-correlation case is given by Eq. 1 when i = j, whereas the dual-color cross-correlation case is given when i ≠ j. Consider two spectrally separated emission signals, a green (Fg) and a red fluorescence signal (Fr), emanating from a mixture of green (g), red (r), and green-red (gr) fluorescent molecules with concentrations Cg, Cr, and Cgr, respectively. The green auto-correlation function Ggg(τ) is derived from Eq. 1 when F = Fj = Fg, the red auto-correlation function Grr(τ) when Fi = Fj = Fr. Cross-correlation between different emission channels Ggr(τ) is derived from Eq. 1 when Fi = Fg and Fj = Fr; it requires red and green fluctuations to arise within a certain sampling time interval which occurs significantly often only when doubly labeled molecules Cgr enter the detection volume. Auto- and cross-correlation functions of signals from freely diffusing fluorophores in ideally calibrated systems that are equally illuminated by two excitation lasers read (18):
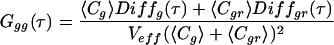 |
2a |
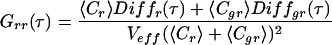 |
2b |
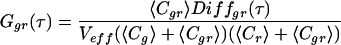 |
2c |
where
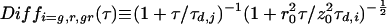 |
3 |
denotes the temporal decay of either correlation function caused by the molecular diffusion of species i with a diffusion time of τd,i. Structural parameters r0 and z0 are determined by the experimental geometry (10) and Veff is the effective measurement volume. Comparing the expressions for auto- and cross-correlation functions in Eqs. 2aa–c, two important differences are obvious (16, 18, 19): (i) the cross-correlation function contains only dynamic information about the doubly labeled species (Diffgr); and (ii) the amplitude (time zero value) of the auto-correlation function, G(0), is inversely proportional to the total concentration of detected fluorophores, whereas the amplitude of the cross-correlation function is directly proportional to the concentration of the doubly labeled species (the denominator containing the fluorescence signals of both emission channels is usually constant during a measurement).
By contemporary analysis of cross- as well as of auto-correlation functions of the system, the number of doubly labeled species can easily be determined at any time from the time zero values as given above. Whatever the reaction either produces or consumes doubly labeled species, absolute product concentrations can be extracted from this expression by calibrating the measurement volume. For τ = 0 in Eqs. 2a and 2b, the denominator of Eq. 2a can be expressed by the single-color auto-correlation amplitudes Ggg (0) and Grr (0), what yields:
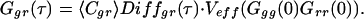 |
4 |
The concentration of molecules bearing both labels, Cgr, is thus given by the fraction of cross-correlation and auto-correlation amplitudes:
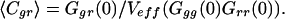 |
5 |
Cross-Correlation Data Analysis and Evaluation.
The experimental cross-correlation curves were fitted with a Marquardt nonlinear least-squares regression routine implemented in the access 2.0 Software (Evotec BioSystems, Hamburg, Germany) by using Eqs. 2a and 3. The structural parameter z0/r0 was determined by evaluating the auto-correlation curves of pure Cy5 and Rhodamine Green solutions. The effective detection volume Veff ≈ 1.33 × 4/3πr02z0 has an ellipsoidal shape where r0 and z0 are the horizontal and vertical axes. Veff was calculated from auto-correlation measurements with a pure Rhodamine Green solution (diffusion coefficient DRhG = 2.8 × 10−10 m2⋅s−1) by using the relationship r0 = (4Dτd)½ (22), which results in an effective detection volume of 0.44 fl. The diffusion time of doubly labeled dsDNA τd,DNA was measured in a reference sample without enzyme. Parameters z0/r0 and τd,DNA were fixed during the analysis of the cross-correlation curves. Concentrations were calculated from the amplitudes obtained from a contemporary cross-correlation and auto-correlation analysis of the reaction samples by using Eq. 5; the value at t∞, which was obtained from a measurement with excess enzyme and which is probably caused by an unspecific detector crosstalk (18), was subtracted as an offset; the resulting values were normalized to the initial substrate concentrations.
RESULTS AND DISCUSSION
A dsDNA substrate containing the GAATTC EcoRI recognition site was employed as a substrate for endonuclease kinetic studies by using dual-color FCS analysis. dsDNA molecules were formed by annealing two synthetic, 66-nt complementary oligonucleotides, which were labeled at their 5′ ends with Cy5 and Rhodamine Green. This pair of fluorescent dyes has been shown previously to be suitable for dual-color cross-correlation experiments (19).
Real-Time Measurements of Endonucleolytic Cleavage.
Cleavage of the double-stranded substrate by EcoRI breaks the chemical linkage between the two different fluorophores resulting in loss of the cross-correlation signal. The time course of this enzyme reaction was monitored in solution by dual-color FCS (Fig. 3). The fluorescence signals were measured continuously, and the cross-correlation analysis was carried out at a rate of one per minute. During the cleavage reaction, the amplitude of the cross-correlation curves corresponding to the concentration of the doubly labeled DNA substrate decreased successively; the second important parameter—the average diffusion time of cross-correlating entities—remained constant, indicating a highly specific detection of the doubly labeled DNA substrate by this method. FCS measurements usually require pico- to nanomolar concentrations of fluorescent molecules for optimal correlation analysis. Here, a kinetic analysis with broader ranges of substrate concentrations was achieved by adding unlabeled substrate to the reaction sample. Cleavage kinetics with different ratios of labeled to unlabeled substrate ranging from 0.05 to 0.5 were obtained by plotting the evaluated substrate concentrations versus time (Fig. 4). These measurements revealed identical kinetics, providing evidence that the fluorophores attached at the ends of the DNA molecule did not interfere with the enzyme’s catalytic action (Fig. 4 Inset); therefore, the labeled substrate served as a one-to-one indicator.
Figure 3.

Cross-correlation curves at different time points during an endonucleolytic cleavage reaction. The 10 nM labeled DNA, the 80 nM unlabeled DNA, and the 1.6 nM EcoRI were incubated in the reaction buffer at 27°C. Dotted lines are the original data, which were fitted with Eqs. 2a and 3. The fitted curves are given in solid lines. During the reaction the cross-correlation amplitude Ggr(0), which is a measure of the reaction progress, gradually decreases.
Figure 4.

Influence of different indicator fractions. Real-time cleavage reactions were monitored at 27°C applying 160 pM EcoRI and 15 nM substrate concentration containing different amounts of labeled substrate (7.5 nM, ▪; 3.0 nM, ○; 1.5 nM, □; 0.75 nM, ○) resulting in fractions ranging from 0.5 to 0.05. (Inset) Evaluated initial slopes were plotted versus fraction values.
Enzyme Kinetics.
The class II restriction endonuclease EcoRI catalyzes the cleavage of the phosphodiester bond between the guanosine and the adenosine monomer in each of both strands of the palindromic recognition sequence GAATTC (21). Different mechanisms have been proposed for the action of class II restriction endonucleases: either the reactions can occur simultaneously on each strand, or the two reactions occur sequentially, with or without the release of an intermediate. Several mechanistic studies have revealed that EcoRI usually cuts both strands sequentially without releasing an intermediate, and that the dissociation of the final product is the rate-limiting step (23, 24), although altered reaction kinetics have been observed that depend on the substrate characteristics as well as on the assay conditions (24–29). The method described in this study detects cleavage reactions by observing the separation of two different fluorescent labels, resulting from scissions in both strands of a single molecule; consequently, the kinetics that is observed refers to an overall reaction rate. Cleavage reactions were monitored over a wide range of substrate concentrations (from 1 to 130 nM) by adding a defined amount of unlabeled substrate to a constant amount of labeled substrate, the latter serving as an indicator. From a linear regression of initial slopes at different substrate concentrations, it was confirmed that EcoRI catalysis obeys the Michaelis–Menten equation (Fig. 5), as already shown by others (30–33). From an Eadie–Hofstee plot (Fig. 5 Inset) a KM of 14 ± 1 nM was derived. As pointed out by McLaughlin et al. (31) the KM value of EcoRI depends sensitively on the size of the substrate. Although most studies cannot be compared due to varying detection methods and reaction conditions (temperature, ionic strength, etc.), plasmid substrates usually revealed KM values between 1 and 10 nM (25, 34–36), whereas the Km was remarkably higher (80–230 nM, in one case up to 7 μM) when short synthetic DNA oligomers were used (30, 31, 33, 37, 38). Compared with these values, the KM obtained here is rather low, and this is most probably caused by the relatively large size of the substrate. Both, the KM as well as the kcat of 4.6 ± 0.2 min−1, confirmed the suitability of dual-color FCS for measuring kinetics in the nanomolar range.
Figure 5.

Michaelis–Menten plot. Labeled DNA at a final concentration of 0.8 nM was mixed with different amounts (0–130 nM) of unlabeled DNA and incubated with 160 pM EcoRI in the reaction buffer at 27°C. The reactions were monitored on-line and the initial rates v were derived by linear regression of data points of the first 5–20 min. (Inset) Calculations from an Eadie–Hofstee plot lead to a KM value of 14 ± 1 nM and vmax of 0.74 ± 0.03 nM min−1.
Quantification of Enzyme Activity.
In addition to the kinetic analyses, the EcoRI cleavage reactions were carried out at various enzyme concentrations to demonstrate the ability of the cross-correlation analysis to quantify sensitively the enzyme activity (Fig. 6). With an initial substrate concentration of 0.8 nM (below the KM) a linear relation between initial reaction rates and enzyme concentrations was obtained over a concentration range of more than one order of magnitude (Fig. 6 Inset). Enzyme activity was detected down to 1.6 pM. There exist several methods for determining the activity of restriction endonucleases; most of them include sampling at different time points and analyzing the cleavage products by gel electrophoreses, chromatography, or affinity separation (33). If linked to radioactive or enzymatic detection, these assays are very sensitive, down to the subnanomolar range, but they suffer from being discontinuous. Methods that monitor enzymatic restriction reactions in real time have been described, either on solid surfaces (39) or in solution, by applying a variety of detection methods, e.g., measurements of increases in absorbance at 254 nm caused by the hyperchromic effect (40), of changes in the fluorescence of ethidium bromide on binding (35, 41), of fluorescence quenching of fluorescein by DNA (42), or of fluorescence resonance energy transfer (FRET) between a donor and an acceptor dye (43). Published data for these methods indicate sensitivities that are at least one order of magnitude below those presented in this work. The extremely low enzyme activities detected by dual-color FCS demonstrate the enormous sensitivity of this technique.
Figure 6.

Reaction rates at different enzyme concentrations. Endonucleolytic reactions were carried out in the reaction buffer at 27°C with 0.8 nM doubly labeled DNA substrate and different enzyme concentrations ranging from 1.6 to 80 pM. Time courses are shown for E0 = 1.6 pM (•), 8 pM (○), and 80 pM (▪). (Inset) Initial rates v were calculated from linear regression of the data points of the first 5–50 min; the plots of these initial rates versus enzyme concentrations E0 indicate a strong linear relationship between v and E0.
CONCLUSIONS
In this work, a highly sensitive and universal method for monitoring the kinetics of catalyzed cleavage or addition reactions in solution in real time is presented. The method applies the principle of dual-color FCS, which allows the discrimination of the fluctuating signals from single molecules with different fluorescence characteristics in a femtoliter volume element. There are several advantages: the assay is homogeneous, the assay conditions such as temperature and buffer compositions can be adjusted to meet specific requirements, and the assay volume may theoretically be scaled down to the focal volume of femtoliters to carry out minute enzyme assays either in single cells or in defined small volumes of solution. In this study enzyme kinetics were measured sensitively in the subnanomolar range, and enzyme activity was measured in the low picomolar range; both these limits are far below those provided by conventional homogeneous assays. A dsDNA molecule was used as the substrate; however, the assay can in principle be applied to any reaction where the chemical linkage between two fluorophore labels is formed or cleaved during an enzymatic action. In principle, there are no limitations in (i) the size of the substrate molecule or (ii) the distance between both fluorophores, as is required for fluorescence resonance energy transfer or for fluorescence quenching methods; therefore, this method is accessible to a much broader class of substrates. Furthermore, the substrate structure recognized by the active site of an enzyme can remain completely unchanged; therefore, the method displays the true enzyme–substrate molecular interactions, which is important for precise studies of enzyme mechanisms, as well as applications of such an assay in designing novel biomolecules. The method exhibits a large potential in fulfilling the requirements for an effective assay for high-throughput screening (HTS) in small volumes and with high sensitivity. By adjusting the assay time to the specific purpose (20), sufficient accuracy for prescreenings in HTS, as well as high accuracy for the precise characterization of hits that must be further validated, can be achieved with the same technique. If only yes-or-no decisions about the presence of doubly labeled species are required, the sampling time for data acquisition for the analysis of cross-correlation amplitudes can be significantly reduced as shown in the subsequent article (RAPID FCS) (20). The evolutionary selection of biopolymers with respect to their catalytic function among a large population of variants requires the precise determination of features of the molecules which have been only slightly improved. Here, we demonstrate the potential of dual-color FCS analysis to fulfill these demands of evolutionary biotechnology. In conclusion, this new method can be applied as a generic principle to many different fields of catalytic investigations, such as diagnostics, analytical determinations, and screening applications with all kinds of catalytic biopolymers. In addition to kinetic studies and activity determinations, dual-color FCS will find useful applications in HTS and evolutionary biotechnology. Especially for the evolutionary creation and optimization of biomolecules, dual-color FCS has the potential of a universal and highly effective tool.
Acknowledgments
We thank Jan Bieschke for close collaboration and critical reading the manuscript. In addition, we thank Melanie Schäfer and Kathrin Baumert for technical assistance; Klaus Dörre, Dr. Jens Stephan, and Dr. Thorsten Winkler for discussions; and Margitta Clegg for reading the manuscript. This work was supported by the German Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie (Grant No. 0310739), by Evotec BioSystems GmbH, Hamburg, and by the Max Planck Society.
ABBREVIATIONS
- FCS
fluorescence correlation spectroscopy
- dual-color FCS
dual-color fluorescence cross-correlation spectroscopy
- dsDNA
double-stranded DNA
References
- 1.Eigen M, Gardiner W. Pure Appl Chem. 1984;56:967–978. [Google Scholar]
- 2.Eigen M, Rigler R. Proc Natl Acad Sci USA. 1994;91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuster P. Acta Biotechnol. 1996;16:3–17. [Google Scholar]
- 4.Koltermann A, Kettling U. Biophys Chem. 1997;66:159–177. doi: 10.1016/s0301-4622(97)00063-x. [DOI] [PubMed] [Google Scholar]
- 5.Magde D, Webb W W, Elson E L. Phys Rev Lett. 1972;29:705–708. [Google Scholar]
- 6.Ehrenberg M, Rigler R. Chem Phys. 1974;4:390–401. [Google Scholar]
- 7.Elson E L, Magde D. Biopolymers. 1974;13:1–27. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 8.Ehrenberg M, Rigler R. Q Rev Biophys. 1976;9:69–81. doi: 10.1017/s003358350000216x. [DOI] [PubMed] [Google Scholar]
- 9.Rigler R, Mets U, Widengren J, Kask P. Eur Biophys J. 1993;22:169–175. [Google Scholar]
- 10.Kinjo M, Rigler R. Nucleic Acids Res. 1995;23:1795–1799. doi: 10.1093/nar/23.10.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwille P, Oehlenschläger F, Walter N G. Biochemistry. 1996;35:10182–10193. doi: 10.1021/bi960517g. [DOI] [PubMed] [Google Scholar]
- 12.Rauer B, Neumann E, Widengren J, Rigler R. Biophys Chem. 1996;58:3–12. doi: 10.1016/0301-4622(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 13.Oehlenschläger F, Schwille P, Eigen M. Proc Natl Acad Sci USA. 1996;93:12811–12816. doi: 10.1073/pnas.93.23.12811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walter N G, Schwille P, Eigen M. Proc Natl Acad Sci USA. 1996;93:12805–12810. doi: 10.1073/pnas.93.23.12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klingler J, Friedrich T. Biophys J. 1997;73:2195–2200. doi: 10.1016/S0006-3495(97)78251-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwille P, Bieschke J, Oehlenschläger F. Biophys Chem. 1997;66:211–228. doi: 10.1016/s0301-4622(97)00061-6. [DOI] [PubMed] [Google Scholar]
- 17.Maiti S, Haupts U, Webb W W. Proc Natl Acad Sci USA. 1997;94:11753–11757. doi: 10.1073/pnas.94.22.11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwille P, Meyer-Almes F-J, Rigler R. Biophys J. 1997;72:1878–1886. doi: 10.1016/S0006-3495(97)78833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwille P. Ph. D. dissertation. Germany: Göttingen and Technical Univ. Braunschweig; 1996. [Google Scholar]
- 20.Koltermann A, Kettling U, Bieschke J, Winkler T, Eigen M. Proc Natl Acad Sci, USA. 1998;95:1421–1426. doi: 10.1073/pnas.95.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hedgpeth J, Goodman H M, Boyer H W. Proc Natl Acad Sci USA. 1972;69:3448–3452. doi: 10.1073/pnas.69.11.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rigler R, Widengren J. Bioscience. 1990;3:180–183. [Google Scholar]
- 23.Heitman J. BioEssays. 1992;14:445–454. doi: 10.1002/bies.950140704. [DOI] [PubMed] [Google Scholar]
- 24.Modrich P. CRC Crit Rev Biochem. 1982;13:287–323. doi: 10.3109/10409238209114231. [DOI] [PubMed] [Google Scholar]
- 25.Halford S E, Johnson N P, Grinsted J. Biochem J. 1979;179:353–365. doi: 10.1042/bj1790353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forsblom S, Rigler R, Ehrenberg M, Philipson L. Nucleic Acids Res. 1976;3:3255–3269. doi: 10.1093/nar/3.12.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langowski J, Urbanke C, Pingoud A, Maass G. Nucleic Acids Res. 1981;9:3483–3490. doi: 10.1093/nar/9.14.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halford S E, Johnson N P. Biochem J. 1983;211:405–415. doi: 10.1042/bj2110405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halford, S. E. (1983) Trends. Biochem. Sci. 455–460.
- 30.Brennan C A, Van Cleve M D, Gumport R I. J Biol Chem. 1986;261:7270–7278. [PubMed] [Google Scholar]
- 31.McLaughlin L W, Benseler F, Graeser E, Piel N, Scholtissek S. Biochemistry. 1987;26:7238–7245. doi: 10.1021/bi00397a007. [DOI] [PubMed] [Google Scholar]
- 32.Alves J, Urbanke C, Fliess A, Maass G, Pingoud A. Biochemistry. 1989;28:7879–7888. doi: 10.1021/bi00445a050. [DOI] [PubMed] [Google Scholar]
- 33.Jeltsch A, Fritz A, Alves J, Wolfes H, Pingoud A. Anal Biochem. 1993;213:234–240. doi: 10.1006/abio.1993.1415. [DOI] [PubMed] [Google Scholar]
- 34.Halford S E, Johnson N P, Grinsted J. Biochem J. 1980;191:581–592. doi: 10.1042/bj1910581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halford S E, Johnson N P. Biochem J. 1981;199:767–777. doi: 10.1042/bj1990767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jack W E, Terry B J, Modrich P. Proc Natl Acad Sci USA. 1982;79:4010–4014. doi: 10.1073/pnas.79.13.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nordlund T M, Andersson S, Nilsson L, Rigler R, Graslund A, McLaughlin L W. Biochemistry. 1989;28:9095–9103. doi: 10.1021/bi00449a021. [DOI] [PubMed] [Google Scholar]
- 38.Greene P J, Gupta M, Boyer H W, Brown W E, Rosenberg J M. J Biol Chem. 1981;256:2143–2153. [PubMed] [Google Scholar]
- 39.Bier F F, Scheller F W. Biosens Bioelectron. 1996;11:669–674. doi: 10.1016/0956-5663(96)83300-8. [DOI] [PubMed] [Google Scholar]
- 40.Waters T R, Connolly B A. Anal Biochem. 1992;204:204–209. doi: 10.1016/0003-2697(92)90162-z. [DOI] [PubMed] [Google Scholar]
- 41.Friedhoff P, Matzen S E, Meiss G, Pingoud A. Anal Biochem. 1996;240:283–288. doi: 10.1006/abio.1996.0358. [DOI] [PubMed] [Google Scholar]
- 42.Lee S P, Porter D, Chirikjian J G, Knutson J R, Han M K. Anal Biochem. 1994;220:377–383. doi: 10.1006/abio.1994.1353. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh S S, Eis P S, Blumeyer K, Fearon K, Millar D P. Nucleic Acids Res. 1994;22:3155–3159. doi: 10.1093/nar/22.15.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]