

Abstract
Dendritic cells (DC) represent the most powerful professional antigen-presenting cells (APC) in the immune system. The aim of the present study was to analyse, on a single-cell basis by multiparametric flow cytometry with simultaneous four-colour staining and a two-step acquisition procedure, the immunophenotypic profile and cytokine production of DC from 67 normal whole peripheral blood (PB) samples. Two clearly different subsets of HLA-II+/lineage− were identified on the basis of their distinct phenotypic characteristics: one DC subset was CD33strong+ and CD123dim+ (0.16 ± 0.06% of the PB nucleated cells and 55.9 ± 11.9% of all PB DC) and the other, CD33dim+ and CD123strong+ (0.12 ± 0.04% of PB nucleated cells and 44.53 ± 11.5% of all PB DC). Moreover, the former DC subpopulation clearly showed higher expression of the CD13 myeloid-associated antigen, the CD29 and CD58 adhesion molecules, the CD2, CD5 and CD86 costimulatory molecules, the CD32 IgG receptor and the CD11c complement receptor. In addition, these cells showed stronger HLA-DR and HLA-DQ expression and a higher reactivity for the IL-6 receptor α-chain (CD126) and for CD38. In contrast, the CD123strong+/CD33dim+ DC showed a stronger reactivity for the CD4 and CD45RA molecules, whereas they did not express the CD58, CD5, CD11c and CD13 antigens. Regarding cytokine production, our results show that while the CD33strong+/CD123dim+ DC are able to produce significant amounts of inflammatory cytokines, such as IL-1β (97 ± 5% of positive cells), IL-6 (96 ± 1.1% of positive cells), IL-12 (81.5 ± 15.5% of positive cells) and tumour necrosis factor-alpha (TNF-α) (84 ± 22.1% of positive cells) as well as chemokines such as IL-8 (99 ± 1% of positive cells), the functional ability of the CD123strong+/CD33dim+ DC subset to produce cytokines under the same conditions was almost null. Our results therefore clearly show the presence of two distinct subsets of DC in normal human PB, which differ not only in their immunophenotype but also in their functionality, as regards cytokine production.
Keywords: human dendritic cells, phenotype, cytokines, flow cytometry
INTRODUCTION
Dendritic cells (DC) play a crucial role in the immune system since they represent the most potent professional antigen-presenting cells (APC) for the initiation of immune responses. This is related to their widespread localization in all sites of antigen entry, their high expression of immunomodulatory molecules necessary for T cell activation, and their production of cytokines [1–6]. In recent years there has been an increasingly high interest on the study of DC, particularly due to their possible application in immunotherapy. Accordingly, the ability of DC to stimulate primary T lymphocyte and T cell-dependent immune responses may provide opportunities for therapeutic intervention in bone marrow and solid organ transplantation, as well as in autoimmune diseases. In addition, protocols for clinical immunotherapy programmes, targeted on malignant cell antigens or infectious agents, have been designed to exploit DC as a ‘natural adjuvant’ for optimal therapeutic vaccination [7–10]. In spite of the large amounts of information accumulated in past years about DC, many questions remain unanswered on the precise origin, the maturation pathways, the different functional roles and the relationship between the different subpopulations of DC. Difficulties in studying human DC mainly relate to their low frequency in tissues and their heterogeneity [1]. Moreover, no specific markers which could be used to identify definitively all DC and their precursors are currently available [11]. Due to their low frequency, the collection of an adequate number of DC—either by isolation procedures [12–18] or by in vitro generation of DC following culture of CD34+ cells [19–21] or monocytes [22]—has been a prerequisite for their further analysis, including their immunophenotypical and functional characterization. However, such procedures for the enrichment of DC may induce the selection of particular cell subsets as well as changes in both the phenotype and functional characteristics of the initial subsets of DC present in the sample. This, together with the use of different methodological approaches for the enrichment and analysis of DC, has contributed to the existence of conflicting results regarding both the phenotype and functional characteristics of the different subsets of DC [23–25] which have been found in peripheral blood (PB), skin and lymphoid organs [15,26–32]. Recent studies have shown the existence of two clearly different populations of circulating DC which display a different origin [30]. Although it has been shown that both subsets of PB DC display the ability to activate naive T cells [29], to the best of our knowledge no systematic study has been performed in which the immunophenotype and the functional characteristics—as regards cytokine production—of non-isolated human PB DC have been extensively investigated using methodological approaches that avoid the need for pre-enrichment steps for DC and allow the analysis of cytokine production on a single-cell basis.
The aim of the present study was to analyse the immunophenotype and ability to produce cytokines of these two subsets of DC present in human PB from healthy subjects using four-colour stainings analysed at flow cytometry on erythrocyte-lysed whole blood samples. Our results clearly show the existence of two different subsets of circulating DC which not only display a distinct immunophenotype but which are also functionally different in terms of cytokine production.
MATERIALS AND METHODS
Normal PB samples
A total of 67 PB samples from healthy adult subjects were analysed in this study. PB specimens were obtained from the University Hospital of Salamanca Blood Bank. The mean age of these individuals was 31 ± 8 years (range 19–54 years; median 32 years); 41 (61%) were males and 26 (39%) females. All PB samples (approx. 5 ml) were collected in EDTA anticoagulant and immediately processed for the immunophenotypical analysis. Additionally, in a subgroup of 13 individuals another tube containing 5 ml of whole blood was collected using heparin as anticoagulant and immediately processed for the analysis of cytokine production. All samples were obtained with the approval of the local Ethical Committee after informed consent had been given by the donor.
Immunophenotypic analysis of MHC II+/lineage− cells
Whole PB samples (approximately 2 × 106 cells in 100 μl/test) were analysed by direct immunofluorescence using four-colour stainings with MoAbs directly conjugated with the following fluorochromes: FITC, PE, peridin chlorophyll protein (PerCP) and allo-phycocyanin (APC). In all tests, a mixture of either FITC- or PE-conjugated MoAbs directed against T lymphocytes, B lymphocytes, natural killer (NK) cells and monocytes was used: CD3–FITC or PE (Leu-4; Becton Dickinson, San Jose, CA), CD19–FITC or PE (Leu-12; Becton Dickinson), CD56–FITC (clone C5.9; IMICO, Madrid, Spain), CD56–PE (Leu-19; Becton Dickinson) and CD14–FITC or PE (Leu-M3; Becton Dickinson), respectively. Likewise, anti-HLA-DR–PerCP (clone L243; Becton Dickinson) and anti-CD4–APC (Leu-3a; Becton Dickinson) reagents were used in all tests. The surface phenotype of the PB MHC II+/lineage− cells was analysed with the large panels of MoAbs shown in Table 1. Briefly, PB samples were incubated for 15 min at room temperature in the dark, in the presence of 5–20 μl of each of the above-mentioned MoAbs, according to the recommendations of the manufacturers. Afterwards, 2 ml of FACS lysing solution (Becton Dickinson) diluted 1:10 (v/v) in distilled water were added and the samples were incubated for another 10 min under the same conditions mentioned above, in order to lyse the non-nucleated erythrocytes. Afterwards, cells were centrifuged (5 min at 540 g) and the cell pellet was washed twice with 4 ml of PBS. Finally, cells were resuspended in 0.5 ml of PBS until analysed in the flow cytometer. Data acquisition was performed in two consecutive steps (as previously described [33]) on a FACSCalibur flow cytometer (Becton Dickinson) equipped with an argon ion laser and a red diode laser. First, 2 × 104 events/test, corresponding to the whole PB sample, were collected; in a second step, information was stored exclusively for those cells included in a HLA-DR++/CD3−, CD14−, CD19−, CD56− (MHC II+/lineage− cells) live gate. In this step, a minimum of 3 × 105 events from the total PB cellularity were measured, in order to obtain a minimum number of 1000 MHC II+/lineage− cells for further immunophenotypical analysis. For data analysis, the Paint-A-Gate PRO software program (Becton Dickinson) was used. Enumeration of MHC II+/lineage− cells present in each PB sample was performed by means of calculating the proportion of HLA-DR++/lineage− cells stored in the second acquisition step from the total number of PB nucleated cells acquired, after excluding cell debris. Absolute counts were obtained by multiplying the percentage of MHC II+/lineage− cells from the total number of PB nucleated cells by the leucocyte count obtained from a Cell-Dyn 4000 haematological cell counter (Abbott, Santa Clara, CA). For the positivity quantification of each of the markers tested, the mean fluorescence intensity (MFI) obtained as assessed by the mean fluorescence channel (arbitrary units scaled from 0 to 10 000) was used. Based on the isotype-matched negative controls, those markers which displayed MFI values > 5 were considered positive. For dimly expressed antigens, such as CD58, the specificity of the labellings was confirmed by incubating the samples with unlabelled MoAb directed to the same antigen prior to incubation with the fluorochrome-conjugated reagent. In order to check the potential day to day variability, a CD3–FITC/CD4–PE/CD8PerCP-stained normal PB sample was analysed in parallel with each set of samples.
Table 1.
Monoclonal antibodies used for the analysis of the surface phenotype of peripheral blood (PB) MHC II+/lineage− cells
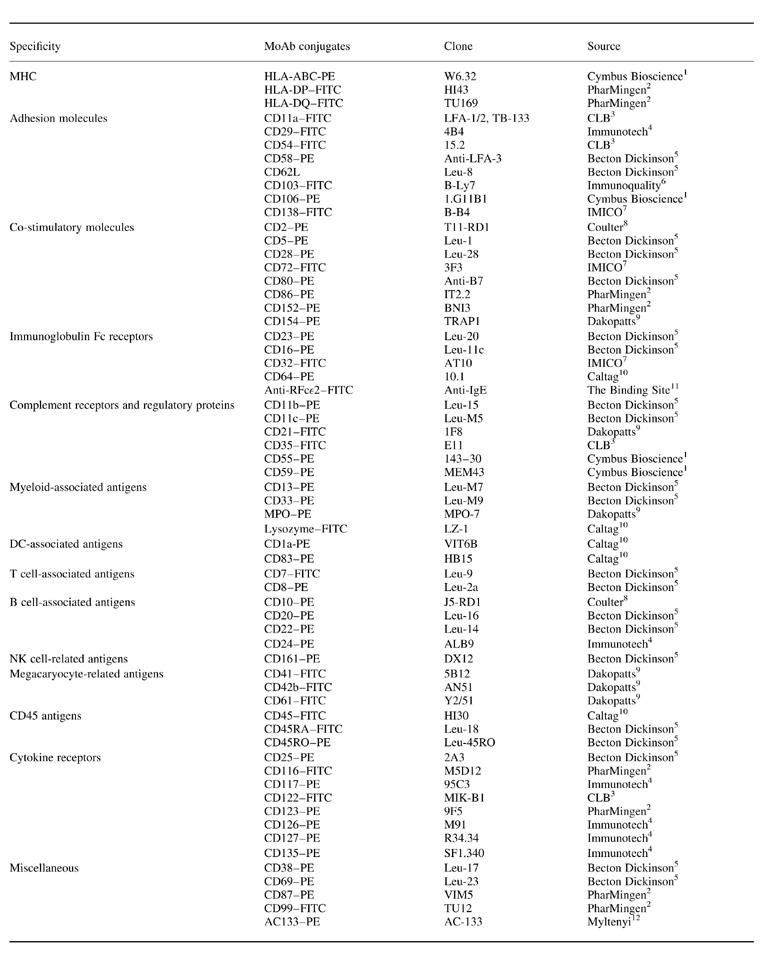
1Cymbus Bioscience, Southampton, UK; 2PharMingen, San Diego, CA; 3CLB, Amsterdam, The Netherlands; 4Immunotech, Marseille, France; 5Becton Dickinson, San Jose, CA; 6Immunoquality Products, Gröningen, The Netherlands; 7IMICO, Madrid, Spain; 8Coulter Corporation, Miami, FL; 9Dakopatts A/S, Glostrup, Denmark; 10Caltag Laboratories, San Francisco, CA; 11The Binding Site, Birmingham, UK; 12Myltenyi, Bergisch Gladbach, Germany.
Analysis of cytokine production by stimulated PB MHC II+/lineage− cells
In a subset of 13 individuals, cytokine production by PB MHC II+/lineage− cells was analysed using a technique that combines four-colour stainings for the specific identification of MHC II+/lineage− cells and the measurement of cytokine production on erythrocyte-lysed whole blood. Briefly, 500 μl of heparin-anticoagulated PB were placed in a tube to which 100 ng/ml of lipopolysaccharide (LPS, from Escherichia coli, serotype 055:B5; Sigma, St Louis, MO) and 10 ng/ml of human recombinant interferon-gamma (IFN-γ; Promega, Madison, WI) were added for the specific stimulation of PB MHC II+/lineage− cells [34]. In addition, 10 μg/ml of brefeldin A (BFA; Sigma) were added in order to block cytokine secretion from cytokine-producing MHC II+/lineage− cells. RPMI 1640 culture medium (BioWhittaker, Walkersville, MD) supplemented with 2 mml-glutamine was added in order to obtain a total volume of 1 ml. An unstimulated sample, also containing BFA and processed in an identical way, was used in each case as a negative control. Afterwards, PB samples were incubated for 6 h at 37°C in a 5% CO2 and 95% humidity, sterile environment. Once this incubation period was completed, the sample was aliquoted in different tubes (100 μl/tube). Then, 20 μl of the CD3–FITC, CD14–FITC, CD19–FITC, CD56–FITC, HLA-DR–PerCP and CD33–APC (Leu-M9; Becton Dickinson) were added to each tube in order to stain the nucleated cells for the specific identification of two different subsets of PB MHC II+/lineage− cells (lineage–FITC−/HLA-DR–PerCP+/CD33–APC++ and lineage–FITC−/HLA-DR–PerCP+/CD33–APC−/+) (Table 2). After gently mixing, cells were incubated for 15 min at room temperature in the dark. Immediately after this incubation period, cells were fixed, permeabilized and stained with MoAb directed against different human cytokines, using the Fix & Perm reagent (Caltag Labs, San Francisco, CA) [35]. The source and specificities of the MoAb reagents used to detect intracytoplasmic human cytokines were as follows: anti-IL-6–PE (clone MQ2–6A3; PharMingen, San Diego, CA), anti-IL-12–PE (clone C11.5, PharMingen), anti-tumour necrosis factor-alpha (TNF-α)–PE (clone Mab11; PharMingen), anti-IL-1β–PE (clone AS10; Becton Dickinson) and anti-IL-8–PE (clone AS14; Becton Dickinson). All cytokine-directed MoAbs were used at saturating concentrations and conditions. Samples stimulated under the same culture conditions in the absence of BFA (n = 5) showed undetectable cytokine levels.
Table 2.
Immunophenotypic characteristics of normal human peripheral blood MHC II+/lineage− cell subpopulations
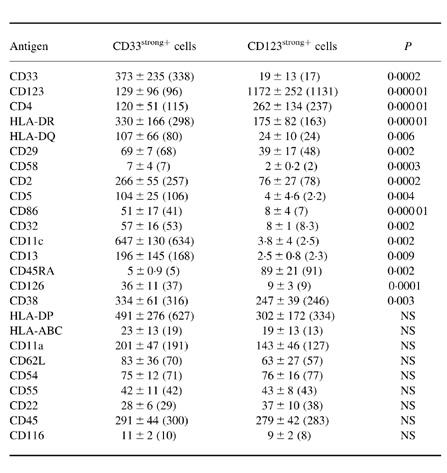
Both MHCII+/lineage− cell populations showed a variable reactivity for the CD59, CD99 and CD135 antigens, and they were constantly negative for the CD1a, CD7, CD8, CD10, CD11b, CD16, CD20, CD21, CD23, CD24, CD25, CD28, CD35, CD41, CD42b, CD61, CD64, CD69, CD80, CD83, CD87, CD103, CD106, CD117, CD122, CD127, CD138, CD152, CD154, CD161, AC133, lysozyme and myeloperoxidase (MPO) antigens.
Results are expressed as mean ±s.d. (median) of mean fluorescence intensity (MFI). NS, Not significant.
For data acquisition and analysis, a FACScalibur flow cytometer was used following the protocol described above. Evaluation of cytokine production was based on both the percentage of positive cells and their MFI, after subtracting the MFI of the negative control.
Isolation of PB dendritic MHC II+/lineage− cells
PB MHC II+/lineage− cells were isolated using magnetic beads according to the Blood Dendritic Cell Isolation Kit protocol from Miltenyi Biotec (Bergisch Gladbach, Germany). Briefly, 500 × 106 nucleated cells were obtained from buffy coat samples according to the conventional procedures of the Blood Bank of the University Hospital of Salamanca. Then, mononuclear cells (MNC) were isolated using Ficoll–Hypaque density gradient centrifugation (density 1077 g/ml; Nyeggard, Oslo, Norway) for 35 min at 250 g. Immediately afterwards, cells were washed twice in PBS supplemented with 0.5% bovine serum albumin (BSA) and 2 mm EDTA (300 g, 10 min) and after the second wash MNC were resuspended in the buffer (300 μl of buffer per 108 cells). In order to deplete the MNC of T lymphocytes, NK cells and monocytes, the samples containing the purified MNC were incubated with 100 μl/108 cells of FcR Blocking Reagent and 100 μl/108 cells of the Hapten-Antibody Cocktail of MoAb (CD3, CD11b, CD16) (10 min at 6–12°C). Cells were then washed (10 min at 300 g) by adding 10 ml of buffer per 108 cells. Once the supernatant had been completely removed, the cell pellet was resuspended in 900 μl of buffer/108 cells and 100 μl of MACS Anti-Hapten Microbeads were added. After an incubation period of 15 min at 6–12°C, cells were passed through a ferromagnetic MACS depletion column (BS or CS type) in a VarioMACS separator (Miltenyi Biotec). Following this step, a positive selection of CD4+ PB MHC II+/lineage− cells was performed. For this purpose the non-magnetic fraction (pre-enriched on MHC II+/lineage− cells) was centrifuged (300 g, 10 min), resuspended in 100 μl of buffer and incubated with 100 μl of MACS CD4 Microbeads (Miltenyi Biotec) for another 30 min at 6–12°C. After washing once by adding 4 ml of buffer (300 g, 10 min), cells were passed twice through a MS+ column in a MiniMACS separator (Miltenyi Biotec) in order to retain CD4+ cells which presumably corresponded to PB DC (MHC II+/lineage− cells).
In order to confirm that the isolated cell fraction was enriched for MHC II+/lineage− cells, cells were immunophenotyped and analysed at flow cytometry. The cocktail of FITC-conjugated MoAbs contained: anti-TCRα/β (clone WT31; Becton Dickinson), anti-TCRγ/δ (clone 11F2; Becton/Dickinson) (in order to exclude T lymphocytes, but not CD3, previously used in the depletion step), CD19, CD56 and CD14. Anti-HLA-DR–PerCP-conjugated was also added. After the isolation procedure, the purity of the sample on MHC II+/lineage− cells was 88%. In addition, the morphological characteristics of the isolated fraction of MHC II+/lineage− cells were assessed. For that purpose, the isolated cell fraction was cytospun by means of a Shandon cytocentrifuge (Shandon, Southern Products, Sewickly, UK); for each cytospin preparation, 100 μl of PBS containing a minimum of 10 × 104 immunoselected cells were used. Slides were stained with May–Grünwald–Giemsa and examined under light microscopy.
Statistical analysis
Mean values and their s.d. as well as the range and median were calculated for each variable by using the SPSS software program (SPSS 6.1.2 Inc., Chicago, IL). The statistical significance of the differences observed between groups for continuous variables was assessed using non-parametric tests (Mann–Whitney U-test). P < 0.01 was considered to be statistically significant.
RESULTS
Identification and enumeration of normal human PB MHC II+/lineage− cells
MHC II+/lineage− cells were identified as cells that lacked markers for T lymphocytes (CD3), B lymphocytes (CD19), NK cells (CD56) and monocytes (CD14) (lineage− cells) and were positive for HLA-DR (strong reactivity) and CD4. The presence of ‘dendritic’ processes in some of MHC II+/lineage− cells (Fig. 1) was confirmed by sorting experiments (immunomagnetic selection) as described previously in Material and Methods. At flow cytometry these cells displayed a typical light scatter pattern, with FSC/SSC values higher than lymphocytes and lower than monocytes (Fig. 2).
Fig. 1.

Morphological characteristics of human peripheral blood MHC II+/lineage− cells after immunomagnetic isolation (May–Grünwald–Giemsa, × 1000). Dendritic processes can only be clearly seen in some of the isolated cells, while others, even lacking cytoplasmic veils, show irregular nuclei shapes.
Fig. 2.
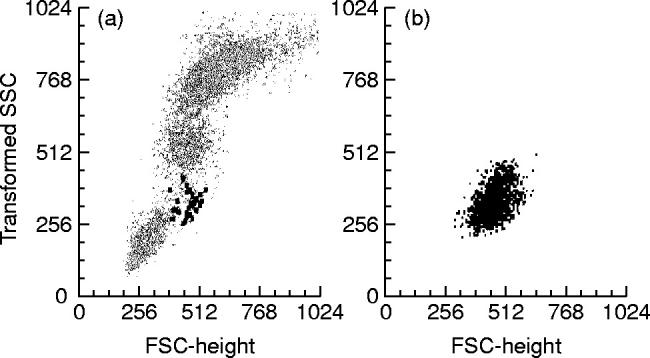
Light scatter characteristics of normal human peripheral blood (PB) MHC II+/lineage− cells (black). (a) All nucleated cells present in PB are shown. (b) Only the (CD3−, CD14−, CD19−, CD56−)/HLA-DR+ gated cells (MHC II+/lineage− cells) from the same PB sample are displayed.
The overall frequency of MHC II+/lineage− cells in the normal PB samples analysed was 0.28 ± 0.08% (range 0.10–0.42%; median 0.29%) of all nucleated cells. The mean absolute number of MHC II+/lineage− cells in the PB was 16 ± 5 DC/μl, ranging from 6 to 34 DC/μl. No statistically significant differences in the number of PB MHC II+/lineage− cells were found with regard either to sex or to the age of the individuals included in the study.
Immunophenotypical identification and characterization of subsets of PB MHC II+/lineage− cells
In all PB samples analysed, two clearly different subsets of MHC II+/lineage− cells were identified on the basis of their distinct phenotypic characteristics. The most widely represented subset of MHC II+/lineage− cells constituted 0.16 ± 0.06% (range 0.023–0.29%; median 0.15%) of all PB nucleated cells and 55.9 ± 11.9% (range 22.9–81.7%; median 58%) of all PB MHC II+/lineage− cells, while the remaining MHC II+/lineage− cell subpopulation represented 0.12 ± 0.04% (range 0.049–0.25%; median 0.11%) of PB nucleated cells and 44.53 ± 11.5% (range 26–77%; median 42%) of all PB MHC II+/lineage− cells. Both subsets of MHC II+/lineage− cells displayed a phenotype in common, characterized by the co-expression of HLA-DR and CD4 in the absence of the CD3, CD19, CD56 and CD14 antigens, which have been associated with other cell lineages. In contrast, these two subsets of PB MHC II+/lineage− cells clearly showed different reactivities for the CD33 and CD123 antigens, the former subset being CD33strong+ and CD123dim+ while the latter was CD33dim+ and CD123strong+ (Fig. 3). Table 2 shows the immunophenotypic characteristics of both subsets of PB MHC II+/lineage− cells according to all the markers analysed in the present study. As may be seen in Table 2, the subpopulation of CD33strong+/CD123dim+ MHC II+/lineage− cells showed a stronger reactivity for the HLA-DR, HLA-DQ, CD29, CD2, CD86, CD32, CD126 and CD38 antigens. Moreover, it was positive for CD58, CD5, CD11c and CD13 and negative for CD45RA. By contrast, CD123strong+/CD33dim+ MHC II+/lineage− cells were CD45RA+, displayed a stronger reactivity for CD4 but were negative for CD58, CD5, CD11c and CD13. Figure 4 illustrates some of these phenotypic differences. For the remaining antigens explored, both subsets of PB MHC II+/lineage− cells showed a similar reactivity. Accordingly, they were constantly positive for HLA-DP, HLA-ABC, CD11a, CD62L, CD54, CD55, CD22, CD45 and CD116, and negative for the CD1a, CD7, CD8, CD10, CD11b, CD16, CD20, CD21, CD23, CD24, CD25, CD28, CD35, CD41, CD42b, CD61, CD64, CD69, CD80, CD83, CD87, CD103, CD106, CD117, CD122, CD127, CD138, CD152, CD154, CD161, AC133, lysozyme and myeloperoxidase (MPO) antigens. In addition, both subsets of PB MHC II+/lineage− cells showed a variable reactivity for the CD59, CD99 and CD135 antigens. Finally, in 50% of PB samples the CD33strong+/CD123dim+ MHC II+/lineage− cell subpopulation showed a heterogeneous expression (from negative to dim positive) for CD45RO and CD72, while both markers were constantly negative in the CD123strong+/CD33dim+ MHC II+/lineage− cell subset.
Fig. 3.
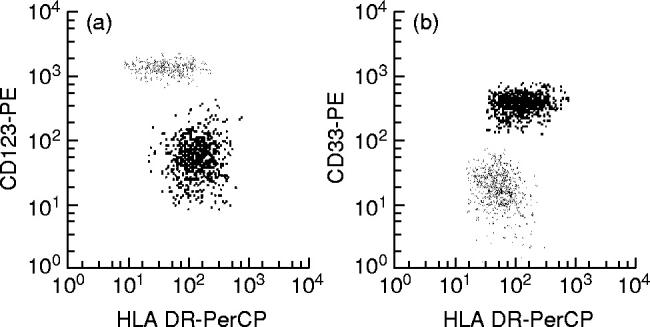
Phenotypic identification of distinct subsets of MHC II+/lineage− cells present in normal human peripheral blood. One MHC II+/lineage− cell population is identified by its strong reactivity for CD123 (grey dots in (a)) and the dim CD33 expression (grey dots in (b)), while the other MHC II+/lineage− subset was CD123dim+ (black dots in (a)) and CD33strong+ (black dots in (b)).
Fig. 4.

Illustrative dot plots of the phenotypic differences between the two peripheral blood (PB) MHC II+/lineage− cell subpopulations. CD33strong+/CD123dim+ MHC II+/lineage− cells are shown in black and CD123strong+/CD33dim+ MHC II+/lineage− cells are displayed in grey.
Cytokine production by PB MHC II+/lineage− cells
In order to evaluate accurately the ability of each of the two MHC II+/lineage− cell subsets present in PB to produce cytokines, their distinct reactivity for the CD33 antigen was used as a discriminative parameter. As shown in Table 3, both subsets of PB MHC II+/lineage− cells displayed a clearly different response to LPS plus IFN-γ as regards cytokine production. Accordingly, while most of the CD33strong+ MHC II+/lineage− cells produced significant levels of IL-1β (97 ± 5% positive cells; MFI 25.3 ± 42.9), IL-6 (96 ± 1.1% positive cells; MFI 606.3 ± 520.3), IL-12 (81.5 ± 15.5% positive cells; MFI 322.3 ± 189.4), TNF-α (84 ± 22.1% positive cells; MFI 514.3 ± 351), and IL-8 (99 ± 1% positive cells; MFI 22 ± 3.4), within the CD33dim+ MHC II+/lineage− cell subset the production of cytokines was extremely low, and could be detected in only < 1% of these cells and with a lower fluorescence intensity as assessed by MFI (IL-1β 0.4 ± 0.6% positive cells, MFI 16; IL-6 0.7 ± 1.1% positive cells, MFI 53; IL-12 0.6 ± 1% positive cells, MFI 99.3 ± 39.4; TNF-α 0.24 ± 0.4% positive cells, MFI 136.3 ± 80.4; IL-8 0.7 ± 1% positive cells, MFI 24). Figure 5 shows representative dot plots of the levels of IL-1β, IL-6, IL-12, TNF-α and IL-8 produced by both subsets of PB MHC II+/lineage− cells in the presence of LPS plus IFN-γ.
Table 3.
Cytokine production by the two MHC II+/lineage− cell subsets present in normal human peripheral blood
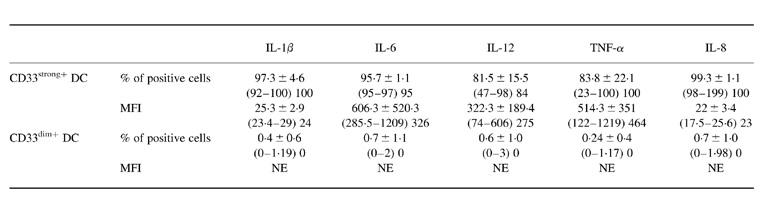
MFI, Mean fluorescence intensity.
Results are expressed as mean ±s.d. (range); median.
NE, Not evaluable due to the small number of positive cells.
Fig. 5.
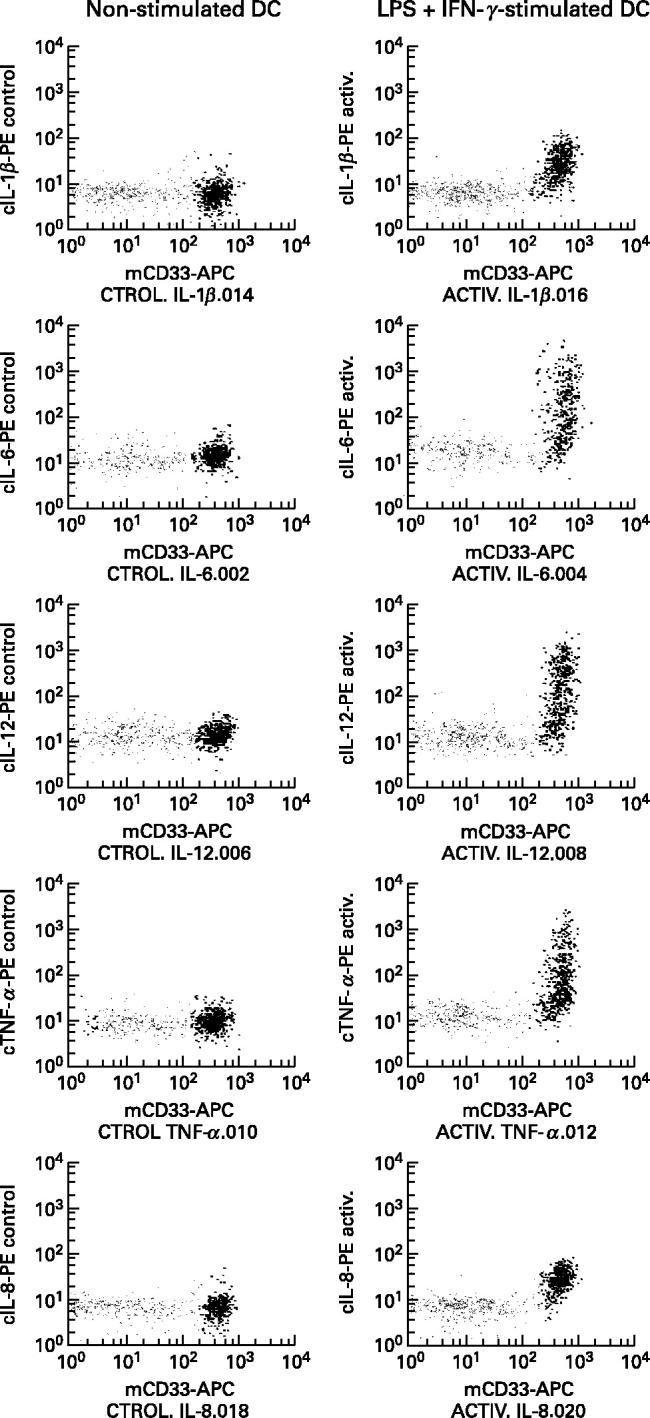
Comparative analysis of cytokine production by the normal human peripheral blood (PB) CD33strong+/CD123dim+ and CD123strong+/CD33dim+ subsets of MHC II+/lineage− cells. The right panels show representative dot plots of IL-1β, IL-6, IL-12, tumour necrosis factor-alpha (TNF-α) and IL-8 production by the CD33strong+/CD123dim+ (black points) and the CD123strong+/CD33dim+ (grey dots) PB MHC II+/lineage− cell subpopulations after stimulation for 6 h in the presence of brefeldin A with lipopolysaccharide (LPS) plus IFN-γ. Left panels show the production of the same cytokines by the two MHC II+/lineage− cell subsets from the same PB sample in the absence of LPS plus IFN-γ stimuli (negative control).
DISCUSSION
Recent studies have shown that DC precursors can be identified in human PB as the fraction of nucleated cells which do not show reactivity for CD3, CD19, CD56 and CD14 antigens and which at the same time are positive for CD4, HLA-DR and other MHC class II molecules [15,21,36]. Moreover, recent studies have shown [15,26,27,30,37] that two different subsets of DC can be identified among the human PB MHC II+/CD4+/lineage− cells, which display distinct phenotypic and functional characteristics. Interestingly, both subsets of human PB DC have been shown to be relatively immature on the basis of their morphology, phenotype and functional behaviour [15,24–27,37–39]. Nevertheless, the definitive relationship between these subsets and the specific functional role of each subpopulation of DC still remains to be established. In spite of the increasing interest in DC, until now no study has simultaneously analysed the immunophenotype of PB DC or their potential immunomodulatory functional role as cytokine-producing cells using a short-term stimulus on DC which have not undergone pre-enrichment steps or other manipulations. Moreover, cytokine production at a single DC level has not been previously analysed in humans.
In the present study we confirm the existence of two clearly distinct subsets of MHC II+/lineage− cells in human PB [15,26,27]. Purification of these cells showed that only a relatively small proportion of them displayed a typical DC morphology or a mature immunophenotype, as reported for the DC present in skin, spleen, Peyer's patches, tonsil and lymph nodes [7,31,40]; however, it should be noted that previous reports have shown that most circulating PB human DC display a characteristic morphology with dendritic processes on their surface only after in vitro culture [39], despite the fact that both cell subsets are able to function as potent APC [15,30]. Olweus et al. [30] and others [15,26,27] have shown that both DC subsets are present in low numbers in normal human PB and that they display clearly different phenotypic features, one subset being CD123strong+/CD33dim+ and the other CD33strong+/CD123dim+. In accordance with these reports, in the present study we show that almost all MHC II+/CD4+/lineage− human PB cells display a characteristic DC phenotype, the frequency of the CD33strong+/CD123dim+ being slightly higher than that of the CD123strong+/CD33dim+ DC subset. Additional phenotypic differences between both subsets of DC were found. Accordingly, while the CD33strong+/CD123dim+ DC population showed a clear expression of myeloid-associated antigens (CD33strong+, CD13strong+), adhesion molecules (CD29, CD58) and costimulatory molecules (CD2, CD5, CD86), together with receptors for IgG (CD32) and complement (CD11c), the other DC subset was characterized by the strong expression of the IL-3α chain receptor (CD123) and a higher reactivity for CD4. These findings would confirm and extend previous observations supporting a more immature origin for the CD123strong+/CD33dim+ DC subset. Accordingly, O'Doherty et al. [26] have reported on the presence of two different subsets of PB DC that could be distinguished on the basis of their different reactivity for the CD11c antigen, one being CD11c+ and the other CD11c−. These authors have shown that while the CD11c− DC are immunologically immature as regards stimulation of allogeneic naive T cells, the CD11c+ DC represent an immunologically mature subset of DC [26]. In a similar way, Thomas et al. [15,27] observed the presence of two different subsets of PB DC based on their reactivity for CD33: CD33strong+ and CD33dim+ DC; the former subpopulation of DC showed reactivity for costimulatory molecules such as CD5 and CD58, as confirmed by our observations.
In spite of the knowledge currently available on both PB DC subsets it remains unclear whether both subpopulations of DC represent different stages of maturation of a unique DC lineage or whether they originate from a different haematopoietic progenitor cell. In this sense, accumulating evidence exists that both subsets of PB cells may relate to different DC maturation pathways that give rise to two different lineages of DC [23,29]. In line with this hypothesis it has been reported in humans that, by culturing cord blood CD34+ haematopoietic progenitor cells in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and TNF-α, two subsets of DC precursors independently emerge [29]. More recently, Olweus et al. [30] suggested that the CD123strong+ DC population represents a separate lineage of cells from that of Langerhans cells and that progenitors that have committed to this CD123strong+ DC lineage are therefore distinct from those that give rise to Langerhans cells when cultured with GM-CSF and TNF-α. In order to gain further insight into these two PB DC subsets, in the present study we have also explored the specific ability of the CD33strong+/CD123dim+ and CD123strong+/CD33dim+ PB DC subpopulations identified to produce cytokines after a short-term in vitro stimulation with LPS plus IFN-γ. Our results show that while CD33strong+/CD123dim+ DC are able to produce significant amounts of inflammatory cytokines such as IL-1β, IL-6, IL-12 and TNF-α and chemokines (IL-8) after a 6-h period when stimulated with LPS plus IFN-γ, the functional ability of the CD123strong+/CD33dim+ DC population to produce cytokines under these conditions is almost nil. To the best of our knowledge this is the first study in which cytokine production by each of these two human PB DC subsets has been specifically investigated at the cytoplasmic level on a single-cell basis. Previous reports have mainly focused on the detection of the production of soluble cytokines after the culture of heterogeneous populations of MNC either enriched or not on DC [34,41].
Overall, our findings regarding cytokine production suggest that both subsets of normal human PB DC may play a different role in priming T cell responses, or alternatively they may correspond to DC in different maturational stages, the CD123strong+/CD33dim+ corresponding to a less differentiated population. However, it may also happen that the use of the combination of LPS plus IFN-γ could be a potent stimulus for cytokine production on CD33strong+/CD123dim+ PB DC while it fails to induce the switch of cytokine secretion on CD123strong+/CD33dim+ DC. In this context, it should be noted that the use of other stimulatory conditions as regards the stimulant used (LPS, phorbol myristate acetate (PMA), ionomycin, IFN-γ), the duration of the stimulus (12–24 h) or the doses and type of cytokine-secretion blocking agent (monensin, BFA plus monensin at different doses) did not increase the response of either of the two DC subsets as regards cytokine production (data not shown). In fact, no other cytokines appeared to have been produced by CD123strong+/CD33dim+ DC, under any of the stimulation conditions described above. In line with our findings, Rissoan et al. [37] have recently shown that following activation of DC through the CD40/CD40L pathway for a period of 1 and 6 days, CD4+/CD11c+ human PB DC produce significant amounts of IL-12, IL-1β, IL-6 and IL-8, while CD4+/CD11c− human PB DC fail to secrete significant amounts of all these cytokines except IL-8.
In conclusion, our results clearly confirm the presence of two distinct subsets of DC in the PB of normal individuals, which display clearly different immunophenotypic features: one associated with more mature characteristics as defined by a greater expression of functional molecules, including costimulatory proteins, as well as IgG and complement receptors. From a functional point of view, we show that both subsets of DC also display a completely different behaviour as regards cytokine production, the CD33strong+/CD123dim+ DC population producing high amounts of the IL-1β, IL-6, IL-12, TNF-α and IL-8 cytokines and the other DC subset almost failing to produce any of the cytokines analysed.
Acknowledgments
This work has been partially supported by a grant (PM 97–0161) from the Dirección General de Ensen~anza Superior (DGES) from the Ministerio de Educación y Cultura (Madrid, Spain) and a grant from the Fondo de Investigaciones Sanitarias de la Seguridad Social (FIS) del Ministerio de Sanidad y Consumo (FIS 99/1239). C.B. is supported by a grant from the Dirección General de Universidades e Investigación (Consejería de Educación y Cultura, Junta de Castilla y León; Valladolid, Spain). J.M.V. and M.L.S. are supported by a grant from Fondos Feder (1FD97–0451) from the DGES, Madrid, Spain.
REFERENCES
- 1.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–96. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 2.Young JW, Steinman RM. Dendritic cells stimulate primary human cytolytic lymphocyte responses in the absence of CD4+ helper T cells. J Exp Med. 1990;171:1315–32. doi: 10.1084/jem.171.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knight SC, Stagg AJ. Antigen-presenting cell types. Curr Opin Immunol. 1993;5:374–82. doi: 10.1016/0952-7915(93)90056-x. [DOI] [PubMed] [Google Scholar]
- 4.Paglia P, Girolomoni G, Robbiati F, Franucci F, Ricciardi-Castagnoli P. Immortalized dendritic cell line fully competent in antigen presentation initiates primary T cell responses in vivo. J Exp Med. 1993;178:1893–901. doi: 10.1084/jem.178.6.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levin D, Constant S, Pasqualini T, Flavell R, Bottomly K. Role of dendritic cells in the priming of CD4+ T lymphocytes to peptide antigen in vivo. J Immunol. 1993;151:6742–50. [PubMed] [Google Scholar]
- 6.Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr Opin Immunol. 1997;9:10–16. doi: 10.1016/s0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- 7.Hart DNJ. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90:3245–87. [PubMed] [Google Scholar]
- 8.Hsu FJ, Benike C, Fagnoni F, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nature Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 9.Choudhury A, Gajewski JL, Liang JC, et al. Use of leukemic dendritic cells for the generation of antileukemic cellular cytotoxicity against Philadelphia chromosome-positive chronic myelogenous leukemia. Blood. 1997;89:1133–42. [PubMed] [Google Scholar]
- 10.Steinman RM. Dendritic cells and immune-based therapies. Exp Hematol. 1996;24:859–62. [PubMed] [Google Scholar]
- 11.O'Doherty UWJ, Swiggard WJ, Inaba Y, et al. Tolerizing mice to human leukocytes: a step toward the production of monoclonal antibodies specific for human dendritic cells. Adv Exp Med Biol. 1993;329:165–75. doi: 10.1007/978-1-4615-2930-9_28. [DOI] [PubMed] [Google Scholar]
- 12.Xu H, Friendrichs U, Gieseler RKH, Ruppert J, Ocklind G, Peters JH. Human blood dendritic cells exhibit a distinct T-cell-stimulatory mechanism and differentiation pattern. Scand J Immunol. 1992;36:689–96. doi: 10.1111/j.1365-3083.1992.tb03129.x. [DOI] [PubMed] [Google Scholar]
- 13.Freudenthal PS, Steinman RM. The distinct surface of human blood dendritic cells as observed after an improved isolation method. Proc Natl Acad Sci USA. 1990;87:7698–702. doi: 10.1073/pnas.87.19.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams LA, Egner W, Hart DN. Isolation and function of dendritic cells. Int Rev Cytol. 1994;153:41–103. doi: 10.1016/s0074-7696(08)62188-9. [DOI] [PubMed] [Google Scholar]
- 15.Thomas R, Davis LS, Lipsky PE. Isolation and characterization of human peripheral blood dendritic cells. J Immunol. 1993;150:821–34. [PubMed] [Google Scholar]
- 16.Egner W, Andreesen R, Hart DNJ. Allostimulatory cells in fresh human blood: heterogeneity in antigen-presenting cell populations. Transplantation. 1993;56:945–50. doi: 10.1097/00007890-199310000-00032. [DOI] [PubMed] [Google Scholar]
- 17.Macey MG, McCarthy DA, Vogiatzy D, Brown KA, Newland AC. Rapid flow cytometric identification of putative CD14− and CD64− dendritic cells in whole blood. Cytometry. 1998;31:199–207. doi: 10.1002/(sici)1097-0320(19980301)31:3<199::aid-cyto7>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 18.Zhou LJ, Tedder TF. Human blood dendritic cells selectively express CD83, a member of the immunoglobulin superfamily. J Immunol. 1995;154:3821–35. [PubMed] [Google Scholar]
- 19.Bernhard H, Disis ML, Heimfeld S, Hand S, Gralow JR, Cheever MA. Generation of immunostimulatory dendritic cells from human CD34+ hematopoietic progenitor cells of the bone marrow and peripheral blood. Cancer Res. 1995;55:1099–104. [PubMed] [Google Scholar]
- 20.Strobl H, Riedl E, Scheinecker C, et al. TGF-beta1 promotes in vitro development of dendritic cells from CD34+ hemopoietic progenitors. J Immunol. 1996;157:1499–507. [PubMed] [Google Scholar]
- 21.Strunk D, Egger C, Leitner G, Hanau D, Stingl G. A skin homing molecule defines the Langerhans cell progenitor in human peripheral blood. J Exp Med. 1997;185:1131–6. doi: 10.1084/jem.185.6.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pickl WF, Majdic O, Kohl P, et al. Molecular and functional characteristics of dendritic cells generated from highly purified CD14+ peripheral blood monocytes. J Immunol. 1996;157:3850–9. [PubMed] [Google Scholar]
- 23.Austyn JM, Phil D. Dendritic cells. Current Opin Hematol. 1998;5:3–15. doi: 10.1097/00062752-199801000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Fanger NA, Wardwell K, Shen L, Tedder TF, Guyre PM. Type I (CD64) and type II (CD32) Fcγ receptor-mediated phagocytosis by human blood dendritic cells. J Immunol. 1996;157:541–8. [PubMed] [Google Scholar]
- 25.O'Doherty U, Steinman RM, Peng M, et al. Dendritic cells freshly isolated from human blood express CD4 and mature into typical immunostimulatory dendritic cells after culture in monocyte-conditioned medium. J Exp Med. 1993;178:1067–78. doi: 10.1084/jem.178.3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Doherty U, Peng M, Gezelter S, et al. Human blood contains two subsets of dendritic cells, one immunologically mature and the other immature. Immunology. 1994;82:487–93. [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas R, Lipsky PE. Human peripheral blood dendritic cell subsets. Isolation and characterization of precursor and mature antigen-presenting cells. J Immunol. 1994;153:4016–28. [PubMed] [Google Scholar]
- 28.Weissman D, Li Y, Ananworanich J, et al. Three populations of cells with dendritic morphology exist in peripheral blood, only one of which is infectable with human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1995;92:826–30. doi: 10.1073/pnas.92.3.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caux C, Vanbervliet B, Massacrier C, et al. CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to GM-CSF + TNFα. J Exp Med. 1996;184:695–706. doi: 10.1084/jem.184.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olweus J, BitMansour A, Warnke R, et al. Dendritic cell ontogeny: a human dendritic cell lineage of myeloid origin. Proc Natl Acad Sci USA. 1997;94:12551–6. doi: 10.1073/pnas.94.23.12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nestle FO, Zheng X-G, Thompson CB, Turka LA, Nickoloff BJ. Characterization of dermal dendritic cells obtained from normal human skin reveals phenotypic and functionally distinctive subsets. J Immunol. 1993;151:6535–45. [PubMed] [Google Scholar]
- 32.Kelsall BL, Strober W. Distinct populations of dendritic cells are present in the subepithelial dome and T-cell regions of the murine Peyer's patch. J Exp Med. 1996;183:237–47. doi: 10.1084/jem.183.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Escribano L, Orfao A, Díaz-Agustín B, et al. Indolent systemic mast cell disease in adults: immunophenotypic characterization of bone marrow mast cells and its prognostic implications. Blood. 1998;91:2731–6. [PubMed] [Google Scholar]
- 34.Hilkens CMU, Kalinski P, de Boer M, Kapsenberg ML. Human dendritic cells require exogenous interleukin-12-inducing factors to direct the development of naive T-helper cells toward the Th1 phenotype. Blood. 1997;90:1920–6. [PubMed] [Google Scholar]
- 35.Escribano L, Orfao A, Díaz-Agustín B, et al. Human bone marrow mast cells from indolent systemic mast cell disease constitutively express increased amounts of the CD63 protein on their surface. Cytometry (Comms Clin Cytometry) 1998;34:223–8. doi: 10.1002/(sici)1097-0320(19981015)34:5<223::aid-cyto3>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 36.Young JW, Steinman RM. The hematopoietic development of dendritic cells: a distinct pathway for myeloid differentiation. Stem Cells (Dayt) 1996;14:376–87. doi: 10.1002/stem.140376. [DOI] [PubMed] [Google Scholar]
- 37.Rissoan M-C, Soumelis V, Kadowaki N, et al. Reciprocal control of T helper cell and dendritic cell differentiation. Science. 1999;283:1183–6. doi: 10.1126/science.283.5405.1183. [DOI] [PubMed] [Google Scholar]
- 38.Schuler G, Thurner B, Romani N. Dendritic cells: from ignored cells to major players in T-cell-mediated immunity. Int Arch Allergy Immunol. 1997;112:317–22. doi: 10.1159/000237474. [DOI] [PubMed] [Google Scholar]
- 39.Teunissen MBM, Wormmeester DJ, Kreig SR, et al. Human epidermal Langerhans cells undergo profound morphologic and phenotypic changes during in vitro culture. J Invest Dermatol. 1990;94:166–73. doi: 10.1111/1523-1747.ep12874439. [DOI] [PubMed] [Google Scholar]
- 40.Steinman RM, Pack M, Inaba K. Dendritic cells in the T-cell areas of lymphoid organs. Immunol Rev. 1997;156:25–37. doi: 10.1111/j.1600-065x.1997.tb00956.x. [DOI] [PubMed] [Google Scholar]
- 41.Verhasselt V, Buelens C, Willems F, De Groote D, Haeffner-Cavaillon N, Goldman M. Bacterial lipopolysaccharide stimulates the production of cytokines and the expression of co-stimulatory molecules by human peripheral blood dendritic cells. Evidence for a soluble CD14-dependent pathway. J Immunol. 1997;158:2919–25. [PubMed] [Google Scholar]