

Abstract
We have previously shown that the gold-containing disease-modifying anti-rheumatic drugs, auranofin (AF) and gold sodium aurothiomalate (GSTM) reduce human umbilical vein endothelial cell (HUVEC) adhesion molecule expression and neutrophil (PMN) adherence. AF diminishes E-selectin and intercellular adhesion molecule-1 (ICAM-1) on cytokine-activated HUVEC, while GSTM decreases only E-selectin. Since tight adhesion is critical for PMN to damage EC, we tested whether these drugs modulated human PMN-mediated injury to TNF-α-activated HUVEC in vitro (as measured by 51Cr release). Here we show that TNF-α caused a prominent PMN-mediated cytotoxicity that was dose-dependently reduced when AF and GSTM were added to the assay system. We also found that a potent inhibitor of NF-κ B, pyrrolidine dithiocarbamate (PDTC) in a dose-dependent manner impaired TNF-α-induced cytotoxicity, indicating a role of NF-κ B activation in cytokine-induced endothelial injury. To examine the effects of AF and GSTM on TNF-α-induced NF-κ B activation this was measured in HUVEC nuclear extracts by an electrophoretic mobility shift assay. AF, but not GSTM, decreased TNF-α-induced NF-κ B activation in HUVEC. Thus, in this in vitro model of vasculitis, AF and GSTM dose dependently reduced TNF-α-mediated neutrophil-dependent cytotoxicity for HUVEC, and AF, but not GSTM, inhibited NF-κ B mobilization, thereby providing possible mechanisms for effects of AF and GSTM.
Keywords: anti-rheumatic drugs, gold salts, NF-κ B, cytotoxicity, endothelial cells
INTRODUCTION
The adhesion of circulating leucocytes to vascular endothelium is pivotal for the inflammatory responses in various disorders, e.g. systemic vasculitis and rheumatoid arthritis [1]. The proinflammatory cytokine TNF-α is known to activate endothelial cells to express adhesive and activation molecules for leucocytes [1]. This can lead to an activation of the cytotoxic capacity of polymorphonuclear neutrophils (PMN), which may result in injury to the endothelial cells that is dependent on adhesion molecules [2,3].
We have recently described that the gold-containing disease-modifying anti-rheumatic drugs, auranofin (AF) and gold sodium thiomalate (GSTM) in vitro modulate human umbilical vein endothelial cell (HUVEC) adhesion molecule expression and PMN adherence to HUVEC [4]. AF diminishes E-selectin and intercellular adhesion molecule-1 (ICAM-1) expression on cytokine-activated HUVEC, while GSTM decreases only E-selectin. We also reported that AF and GSTM are both potent inhibitors of endothelial cytotoxicity induced when activated PMN are added [5] and may also reduce endothelial injury when HUVEC have been activated by cytokines and quiescent PMN subsequently added [6]. We and others have shown that AF may inhibit degranulation and chemotaxis of PMN in vitro, and GSTM has been reported to interfere with endothelial proliferation in vitro and synovial expression of E-selectin in vivo[7–10].
Induction of adhesion molecules, e.g. ICAM-1 and E-selectin, by TNF-α and other inflammatory cytokines is regulated at the level of gene transcription and requires the transcription factor nuclear factor-κB (NF-κ B) [11,12]. The NF-κ B/Rel transcription family plays an important role in cytokine-induced gene activation [13]. This family consists of p50 (NFκ B1), p52 (NFκ B2), p65 (RelA), RelB, v-Rel and c-Rel. NF-κ B is maintained in the cytoplasm of cells in an inactive form in association with Iκ B-α. TNF-α stimulation triggers the release of NF-κ B from Iκ B, resulting in NF-κ B translocation to the nucleus, where it binds to DNA at specific κ B sites, rapidly inducing a variety of genes, encoding signalling proteins [12,14–16]. In endothelial cells, the p50-p65 heterodimer is the dominant species that binds to κ B consensus sequences in ICAM-1 and E-selectin genes and activates gene transcription. Thus, agents that block NF-κ B activation have the potential to inhibit inflammatory processes in endothelial cells. The two important anti-inflammatory agents salicylates and glucocorticoids have recently been reported to inhibit NF-κ B-driven gene expression, which may offer an explanation, at least partly, to the anti-inflammatory actions of these drugs [17–19].
Since tight adhesion is critical for PMN to damage EC, we tested whether AF and GSTM modulated human PMN-mediated injury to TNF-α-activated HUVEC in vitro by decreasing TNF-α-induced NF-κ B activation.
MATERIALS and METHODS
Chemicals
Heparin, pyrrolidine dithiocarbamate (PDTC), TNF-α (2 × 107 U/mg) and Triton X-100 were obtained from Sigma Chemical Co. (St Louis, MO). AF was from Smith Kline Beecham (Brentford, UK), and GSTM from Rhone-Poulenc Rorer (Birkeröd, Denmark). 51Cr was from Du Pont Co. (Wilmington, DE), endothelial cell growth factor from Collaborative Research, Inc. (Bedford, MA) and 24-well polystyrene plates (2 cm2/well) from Nunclon (Roskilde, Denmark). Fetal calf serum (FCS), HEPES, penicillin, streptomycin, RPMI 1640 and Hanks’ balanced salt solution (HBSS) were from Gibco (Paisley, UK). Collagenase (type 3) was supplied by Worthington (Freehold, NJ), Sephadex G25 and Percoll by Pharmacia Fine Chemicals (Uppsala, Sweden). EDTA was from Merck (Darmstadt, Germany). All other chemicals were also of the highest purity and quality commercially obtainable.
Endothelial cell cultures
HUVEC were obtained from human umbilical veins by treatment with 0·2% collagenase as described [20]. Cells were suspended in culture medium (RPMI 1640 with 20% FCS, 90 μg heparin/ml, 50 μg endothelial cell growth factor/ml, 100 U penicillin/ml, 100 mg streptomycin/ml, HEPES, sodium pyruvate and non-essential amino acids) and grown in 80-cm2 tissue culture flasks precoated with 2% gelatin. Culture medium was changed the following day and then three times per week. HUVEC were trypsinized when confluent, resuspended in medium and seeded into 24-well plates (2 cm2 per well) and utilized as primary cultures upon achieving confluence.
Preparation of neutrophils
Neutrophils were prepared by a one-step discontinuous Percoll gradient centrifugation on venous blood obtained from healthy volunteers, as described previously [21]. The purified PMN (> 98% purity and viability) were washed twice before lysis of contaminating erythrocytes with ice cold ammonium chloride (0·155 m) and resuspended in HBSS at pH 7·4.
51Cr-release cytotoxicity assay
HUVEC, obtained from human umbilical veins, were grown in tissue culture flasks, resuspended and seeded into 24-well plates and utilized as primary cultures [20]. HUVEC monolayers were labelled with 6 μCi of 51Cr for 24 h, washed and TNF-α (100 ng/ml) was added to the labelled monolayer and dishes were incubated for 24 h at 37°C in a 5% CO2 incubator. After two washes the monolayer was covered with 1·25 × 106 PMN in HBSS with 1% FCS in a final volume of 500 μl, yielding an effector to target (endothelial cell) ratio of 6·25:1. Dishes with HUVEC and PMN were incubated for 4 h at 37°C. When the incubation was terminated supernatants were centrifuged to pellet any HUVEC that may have detached but not lysed. HUVEC monolayers were lysed and the radioactivity of supernatants, pellet fractions and the lysed HUVEC was counted in a gamma counter. The intra-assay variations were small and seldom differed more than ±5% of a mean value.
Nuclear extracts and electrophoretic mobility shift assay
HUVEC were either left untreated or were incubated with AF (1 or 5 μg/ml) or GSTM (50 μg/ml) for 30 min and then stimulated with TNF-α (100 U/ml) for 2 h. Nuclear proteins were extracted in the presence of 1 μg/ml aproptinin, 1 μg/ml antipain, 1 μg/ml chymostatin, 0·1 μg/ml leupeptin and 1 μg/ml pepstatin in order to inhibit proteases, as described previously [22]. A NF-κ B-specific single-stranded ‘anti-sense’ oligonucleotide (5′-CCA GAT GGC CTC TCG GAA AGT CCC CTC TGT TGA G-3′) was filled in labelled using the Klenow fragment of Escherichia coli DNA polymerase 1 in the presence of α32P-dGTP and α32P-dCTP. Electrophoretic mobility shift assay (EMSA) was conducted as previously described [23]. Nuclear extract (10 μg) was incubated with 50 000 ct/min of radiolabelled NF-κ B oligonucleotide at room temperature for 20 min in the presence of 1 μg of poly (dI-dC) and the resulting complexes were separated on a 6% polyacrylamide gel in Tris/borate/EDTA buffer. A 100-fold molar excess of unlabelled oligonucleotide was included in competition experiments. For supershift assays, 2 μg of a polyclonal anti-NF-κ B p50 or p65 subunit antibody were added to the nuclear extract simultaneously with the labelled probe. Autoradiographic signals were quantified by scanning laser densitometry.
Statistical evaluation was performed with Student’s two-tailed t-test for paired samples, when appropriate.
RESULTS
We reported earlier that TNF-α causes a prominent PMN-mediated endothelial cytotoxicity [2]. To investigate the effect of gold-containing disease-modifying anti-rheumatic drugs on the TNF-α-induced neutrophil-dependent endothelial injury we examined whether the drugs protected EC by incubating the HUVEC monolayers with the anti-rheumatic drug for 30 min, washed the monolayers, added TNF-α for 24 h, rinsed and then co-incubated with PMN.
AF as well as GSTM significantly hampered TNF-α-induced endothelial injury (Figs 1 and 2). The dose curves for these two gold salts have been reported [6]. Thus, AF as well as GSTM protect HUVEC from neutrophil-dependent cytotoxicity induced by TNF-α.
Fig. 1.
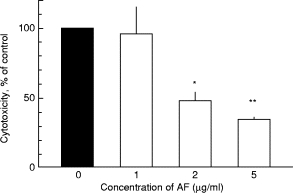
The effect of auranofin (AF) on the cytotoxicity induced by 100 ng/ml of TNF-α. Human umbilical vein endothelial cells (HUVEC) were treated for 30 min with AF, rinsed and then exposed to TNF-α for 24 h; subsequently unstimulated polymorphonuclear neutrophils (PMN) were added. Mean and s.e.m. values for five experiments, run in duplicates. **P < 0001; *P < 0·01 compared with controls, i.e. cytotoxicity induced by TNF-α without AF. No cytotoxicity was found when HUVEC were treated with TNF-α and AF without any PMN present.
Fig. 2.
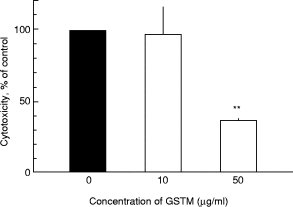
The effect of gold sodium aurothiomalate (GSTM) on the cytotoxicity induced by 100 ng/ml of TNF-α. Human umbilical vein endothelial cells (HUVEC) were treated for 30 min with GSTM, rinsed and then exposed to TNF-α for 24 h; subsequently unstimulated polymorphonuclear neutrophils (PMN) were added. Mean and s.e.m. values for five experiments, run in duplicates.**P < 0·01 compared with controls, i.e. cytotoxicity induced by TNF-α without GSTM. No cytotoxicity was found when HUVEC were treated with TNF-α and GSTM without any PMN present.
Based on our previous findings that expression of the adhesion molecules E-selectin and ICAM-1 on cytokine-activated HUVEC monolayers is essential for the endothelial injury in vitro[2], and since this expression requires the transcription factor NF-κ B, we studied whether a potent inhibitor of NF-κ B, PDTC, might interact with the TNF-α-induced endothelial damage [24,25]. We found that PDTC in a dose-dependent way impaired TNF-α-induced cytotoxicity, thereby indicating a role of NF-κ B activation in cytokine-induced endothelial injury (data not shown).
To examine the effects of gold-containing anti-rheumatic drugs on TNF-α-induced NF-κ B activation we carried out EMSA to determine the levels of NF-κ B in nuclear extracts from HUVEC treated with TNF-α in the presence of AF or GSTM.
As previously described, TNF-α (100 U/ml) induced a nuclear translocation of NF-κ B (Figs 3 and 4) [11]. Competition of the NF-κ B complexes with unlabelled oligonucleotide demonstrated that these complexes were specific for the NF-κ B site (Figs 3 and 4).
Fig. 3.
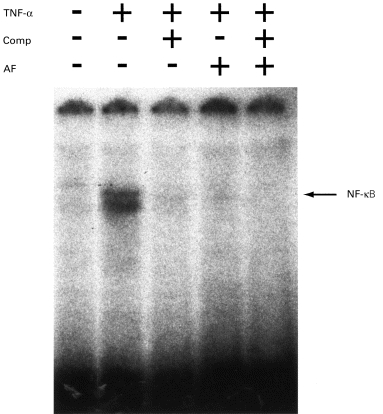
The effect of auranofin (AF) on TNF-α-induced NF-κ B activation. Human umbilical vein endothelial cells (HUVEC) were left untreated (lanes 1–3) or incubated with 1 μg/ml AF for 30 min (lanes 4 and 5). Cells were then stimulated with TNF-α (100 U/ml) for 2 h (lanes 2–5). Reactions were performed in the presence of a 100-fold molar excess of unlabelled NF-κ B consensus oligonucleotide (lanes 3 and 5). Nuclear extracts were analysed subsequently for DNA binding activity in electrophoretic mobility shift assays. The position of the inducible NF-κ B complexes is noted.
Fig. 4.
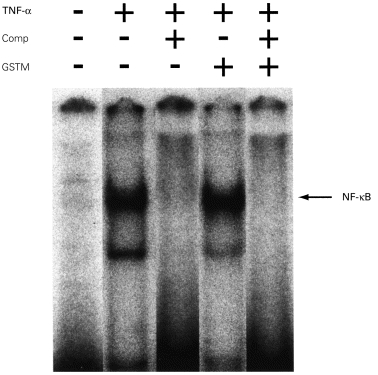
The effect of gold sodium aurothiomalate (GSTM) on TNF-α-induced NF-κ B activation. Human umbilical vein endothelial cells (HUVEC) were left untreated (lanes 1–3) or incubated with 50 μg/ml GSTM for 30 min (lanes 4 and 5). Cells were then stimulated with TNF-α (100 U/ml) for 2 h (lanes 2–5). Reactions were performed in the presence of a 100-fold molar excess of unlabelled NF-κ B consensus oligonucleotide (lanes 3 and 5). Nuclear extracts were analysed subsequently for DNA binding activity in electrophoretic mobility shift assays. The position of the inducible NF-κ B complexes is noted.
When HUVEC were preincubated with AF (1 μg/ml) for 30 min followed by addition of TNF-α for 2 h, AF abolished TNF-α-induced NF-κ B activation in HUVEC (Fig. 3). Similar results were seen with a higher concentration of AF, 5 μg/ml (data not shown). The intensity of the bands was assessed by densitometry and AF reduced the activation of NF-κ B induced by TNF-α by 94% (data not shown).
However, when the cells were pretreated with GSTM (50 μg/ml) for 30 min followed by incubation with TNF-α for 2 h, no significant effect on the TNF-α-induced NF-κ B activation was observed (Fig. 4).
DISCUSSION
This study indicates that AF and GSTM are potent inhibitors of cytokine-mediated neutrophil-dependent cytotoxicity for HUVEC in vitro, and that AF, but not GSTM, impaired NF-κ B mobilization. The two gold-containing disease-modifying anti-rheumatic drugs studied here have both gained general acceptance as effective therapies for RA.
In vitro, injury to endothelial cells by PMN can be induced either by directly activating the neutrophils [20] or by activation of EC by means of cytokines. The mechanisms for inducing cytotoxicity in either system as well as the effects of anti-rheumatic drugs have common but also separate features. Thus, we have previously shown that IL-1β, interferon-gamma (IFN-γ) and TNF-α are powerful inducers of PMN-mediated cytotoxicity for HUVEC in vitro[2]. This cytotoxic effect of IL-1β, IFN-γ and TNF-α is dependent on PMN, the expression of E-selectin and ICAM-1 on HUVEC, associated with nitric oxide (NO) generation in the system, possibly involving PMN activation via a surface receptor and phospholipase C-mediated events, but unrelated to the release of granule constituents and of oxygen radicals [2]. These findings suggest that adhesive interactions between PMN and HUVEC are of significance for the cytotoxic process and that drugs attenuating this process might be anti-inflammatory.
Heimburger et al. [4] recently demonstrated that AF and GSTM may modulate PMN adherence and HUVEC adhesion molecule expression. AF as well as GSTM reduced IL-1β-induced endothelial hyperadhesiveness for PMN, AF decreased E-selectin and ICAM-1 expression on cytokine-activated HUVEC, while GSTM diminished only E-selectin [4]. Thus, the two gold salts appear to interact with HUVEC and EC in discrete ways.
Further evidence for separate effects of AF and GSTM is provided here. As we have previously shown [6], AF and GSTM hampered HUVEC activation by IL-1β and TNF-α. Addition of AF, but not GSTM, reduced the high levels of IL-8 produced by TNF-α-treated HUVEC [6]. IL-8 is a neutrophil chemokine and up-regulator of membrane adhesion receptors in neutrophils [26,27] and the IL-8 gene has been reported being co-operatively regulated by both NF-κ B and CEBP/B transcription factors [28]. In addition, AF and GSTM protected HUVEC from injury by PMN in a dose-dependent manner. The latter effect might be related to increased HUVEC resistance towards NO generated in the system [6]. Thus, AF and GSTM hamper cytokine-induced neutrophil-dependent endothelial injury by effects on the PMN as well as on HUVEC.
In this study we found that a well-known potent inhibitor of NF-κ B [24,25], PDTC, in a dose-dependent manner reduced TNF-α-induced cytotoxicity, indicating a role of NF-κ B activation in cytokine-induced endothelial injury. However, the exact molecular mechanisms through which PDTC could elicit cell signaling effects in a cellular setting remain unclear. The mechanisms for the effects of PDTC on NF-κ B are likely to involve inhibition of binding of the transcription factor to DNA rather than an effect on the activation process [29]. The use of adenoviral gene transfer of the Iκ B-α molecule in EC, which ensures a specific inhibition of NF-κ B, implies that ICAM-1 and vascular cell adhesion molecule-1 (VCAM-1) are under NF-κ B control [30]. We also observed that AF, but not GSTM, inhibited NF-κ B mobilization. This may offer an explanation, at least partly, to the anti-inflammatory actions of AF and why GSTM only reduces E-selectin expression on TNF-α-activated HUVEC, while AF decreases both E-selectin and ICAM-1. The fact that AF is a NF-κ B inhibitor has been suggested in earlier studies [31,32], and there have also been observations indicating that AF but not GSTM inhibits the induction of TNF-α in macrophages [33,34].
Recently it has become clearer that many clinically important anti-inflammatory agents, including salicylates, glucocorticoids and NO, share the ability to abolish NF-κ B-driven gene expression in both leucocytes and endothelial cells [17–19,35]. Salicylates were suggested to inhibit activation of NF-κ B by preventing phosphorylation and subsequent degradation of the inhibitor Iκ B-α[17]. The synthetic glucocorticoid dexamethasone induced the transcription of the Iκ B-α gene, which resulted in an increased rate of Iκ B-α protein synthesis and markedly reduced the amount of NF-κ B that translocated to the nucleus [19]. So far, it is not known whether there is an effect of AF on Iκ B-α production or degradation.
One might also consider other (in)activation pathways for NF-κ B. Reactive oxygen mediators may act as regulators of NF-κ B [36] in EC, with a tyrosine kinase-dependent mechanism [37]. A number of anti-oxidants have been reported to inhibit cytokine-induced nuclear translocation of NF-κ B and induced Iκ B-α phosphorylation [38,39]. Since AF can inhibit fMLP-induced release of superoxide, H2O2 and granule contents, as well as cytosolic calcium transients, in PMN [40,41] it is also possible that AF inhibits NF-κ B translocation by acting as anti-oxidant to affect the NF-κ B/Iκ B-α signalling pathway. Furthermore, AF might also act directly to block kinase cascades involved in TNF-α signalling of NF-κ B.
Thus, in this in vitro model of vasculitis AF and GSTM inhibit TNF-α-induced neutrophil-dependent endothelial injury. AF, but not GSTM, abolished NF-κ B mobilization. These new findings can help in understanding some of the suppressive effects of these disease-modifying anti-rheumatic drugs on rheumatoid vasculitis and inflammation. The clinical importance of AF and GSTM as anti-rheumatic agents may result from the ability to inhibit the expression of adhesion molecules involved in leucocyte recruitment. It is anticipated that novel effective inhibitors of NF-κ B will be identified and these may hold promise as useful immunosuppressive and anti-inflammatory agents.
Acknowledgments
This study was supported by the Swedish Medical Research Council (19X-05991, 19P-8884), the Funds of the Karolinska Institute, King Gustaf V’s 80-year Fund, the Funds of the Swedish Medical Association, Börje Dahlin and Professor Nanna Svartz, Swedish Heart and Lung Foundation and the Swedish Association Against Rheumatism. The skilful technical assistance of Mrs Anette Landström and Mr Christer Forsbom is gratefully acknowledged.
REFERENCES
- 1.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–101. [PubMed] [Google Scholar]
- 2.Bratt J, Palmblad J. Cytokine induced neutrophil mediated injury of human endothelial cells. J Immunol. 1997;159:912–8. [PubMed] [Google Scholar]
- 3.Westlin WF, Gimbrone MA. Neutrophil-mediated damage to human vascular endothelium: role of cytokine activation. Am J Pathol. 1993;142:117–28. [PMC free article] [PubMed] [Google Scholar]
- 4.Heimburger M, Lerner R, Palmblad J. Effects of antirheumatic drugs on adhesiveness of endothelial cells and neutrophils. Biochem Pharmacol. 1998;56:1661–9. doi: 10.1016/s0006-2952(98)00201-9. [DOI] [PubMed] [Google Scholar]
- 5.Bratt J, Palmblad J. Inhibition of neutrophil dependent cytotoxicity for human endothelial cells by antirheumatic drugs. J Lab Clin Med. 1996;128:552–60. doi: 10.1016/s0022-2143(96)90127-4. [DOI] [PubMed] [Google Scholar]
- 6.Bratt J, Palmblad J. Effects of antirheumatic drugs on cytokine induced neutrophil dependent cytotoxicity for human endothelial cells. 1999 Submitted. [PubMed] [Google Scholar]
- 7.Hafström I, Udén AM, Palmblad J. Modulation of neutrophil functions by auranofin. Scand J Rheumatol. 1983;12:97–105. doi: 10.3109/03009748309102893. [DOI] [PubMed] [Google Scholar]
- 8.Finkelstein AE, Roisman FR, Ladizesky MG, Walz DT. Auranofin and lysosomal enzymes. J Rheumatol. 1982;8:46–53. [PubMed] [Google Scholar]
- 9.Kawakami A, Eguchi K, Migita K, et al. Inhibitory effects of gold sodium thiomalate on the proliferation and interferon-γ induced HLA-DR expression in human endothelial cells. J Rheumatol. 1990;17:430–5. [PubMed] [Google Scholar]
- 10.Corkill MM, Kirkham BW, Haskard DO, Barbatis C, Gibson T, Panayi GS. Gold treatment of rheumatoid arthritis decreases synovial expression of the endothelial leukocyte adhesion receptor ELAM-1. J Rheumatol. 1991;18:1453–60. [PubMed] [Google Scholar]
- 11.Read M, Whitley M, Williams A, Collins T. NF-κB and IκB-α: an inducible regulatory system in endothelial activation. J Exp Med. 1994;179:503–10. doi: 10.1084/jem.179.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins T, Read MA, Neal AS, Whitley M, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-κB and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 13.Thanos D, Maniatis T. NF-κB: a lesson in family values. Cell. 1995;80:529–32. doi: 10.1016/0092-8674(95)90506-5. [DOI] [PubMed] [Google Scholar]
- 14.Beg A, Finco T, Nantermet P, Baldwin A. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IκB-α: a mechanism for NF-κB activation. Mol Cell Biol. 1993;15:3301–10. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brockman J, Schierer D, McKinsey T, Hall S, Qi X, Lee W, Ballard D. Coupling of a signal response domain in IκB-α: a mechanism for NF-κB activation. Mol Cell Biol. 1995;15:2809–18. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henkel T, Machleidt T, Alkalay I, Kronke M, Ben-Neriah Y, Baeuerle P. Rapid proteolysis of IκB-α is necessary in the activation of transcription factor NF-κB. Nature. 1993;365:182–5. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- 17.Pierce JW, Read MA, Ding H, Luscinskas FW, Collins T. Salicylates inhibit IκB-α phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration. J Immunol. 1996;156:3961–9. [PubMed] [Google Scholar]
- 18.Kopp E, Ghosh S. Inhibition of NF-κB by sodium salicylate and aspirin. Science. 1994;265:956–9. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 19.Auphan N, Didonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science. 1995;270:286–90. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 20.Bratt J, Lerner R, Ringertz B, Palmblad J. Lipoxin A4 induces neutrophil dependent cytotoxicity for human endothelial cells. Scand J Immunol. 1994;39:351–4. doi: 10.1111/j.1365-3083.1994.tb03385.x. [DOI] [PubMed] [Google Scholar]
- 21.Ringertz B, Palmblad J, Lindgren JÅ. Stimulus-specific neutrophil aggregation: evaluation of possible mechanisms for the stimulus-response apparatus. J Lab Clin Med. 1985;106:132–40. [PubMed] [Google Scholar]
- 22.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–16. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 24.Bessho R, Matsubara K, Kubota M, et al. Pyrrolidine dithiocarbamate, a potent inhibitor of nuclear factor kB (NF-κB) activation, prevents apoptosis in human promyelocytic leukemia HL-60 cells and thymocytes. Biochem Pharma. 1994;48:1883–9. doi: 10.1016/0006-2952(94)90586-x. [DOI] [PubMed] [Google Scholar]
- 25.Ferran C, Millan MT, Csizmadia V, Cooper JT, Brostjan C, Bach FH, Winkler H. Inhibition of NF-κB by pyrrolidine dithiocarbamate blocks endothelial cell activation. Biochem Biophys Res Commun. 1995;214:212–23. doi: 10.1006/bbrc.1995.2277. [DOI] [PubMed] [Google Scholar]
- 26.Baggiolini M, Walz A, Kunkel SL. Neutrophil activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest. 1989;84:1045–9. doi: 10.1172/JCI114265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Detmers PA, Lo SK, Olsen-Egbert E, Walz A, Baggiolini M, Cohn ZA. Neutrophil-activating protein-1/interleukin-8 stimulates the binding activity of the leukocyte adhesion receptor CD11b/CD18 on human neutrophils. J Exp Med. 1990;171:1155–62. doi: 10.1084/jem.171.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunsch C, Rosen CA. NF-κB subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. 1993;13:6137–46. doi: 10.1128/mcb.13.10.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moellering D, McAndrew J, Jo H, Darley-Usmar VM. Effects of pyrrolidine dithiocarbamate on endothelial cells: protection against oxidative stress. Free Rad Biol Med. 1999;26:1138–45. doi: 10.1016/s0891-5849(98)00300-1. [DOI] [PubMed] [Google Scholar]
- 30.Wrighton CJ, Hofer-Warbinek R, Moll T, Eytner R, Bach FH, de Martin R. Inhibition of endothelial cell activation by adenovirus-mediated expression of I kappa B alpha, an inhibitor of the transcription factor NF-kappa B. J Exp Med. 1996;183:1013–22. doi: 10.1084/jem.183.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang JP, Merin JP, Nakano T, Kato T, Kitade Y, Okamoto T. Inhibition of the DNA binding activity of NF-kappa B by gold compounds in vitro. Febs Letters. 1995;361:89–96. doi: 10.1016/0014-5793(95)00157-5. [DOI] [PubMed] [Google Scholar]
- 32.Daniel LW, Civoli F, Rogers MA, Smitherman PK, Raju PA, Roederer M. ET-18-OCH3 inhibits nuclear factor-kappa B activation by 12-O-tetradecanoylphorbol-13-acetate but not by tumor necrosis factor-alpha or interleukin 1 alpha. Cancer Res. 1995;55:4844. [PubMed] [Google Scholar]
- 33.Danis VA, Franic GM, Brooks PM. The effect of slow-acting anti-rheumatic drugs (SAARDs) and combinations of SAARDs on monokine production in vitro. Drugs Exp Clin Res. 1991;17:549–54. [PubMed] [Google Scholar]
- 34.Bondeson J, Sundler R. Auranofin inhibits the induction of interleukin 1 beta and tumor necrosis factor alpha mRNA in macrophages. Biochem Pharmacol. 1995;50:1753–9. doi: 10.1016/0006-2952(95)02030-6. [DOI] [PubMed] [Google Scholar]
- 35.Peng HB, Libby P, Liao JK. Induction and stabilization of IκBa by nitric oxide mediates inhibition of NF-κB. J Biol Chem. 1995;270:14214–9. doi: 10.1074/jbc.270.23.14214. [DOI] [PubMed] [Google Scholar]
- 36.Dalton TP, Shertzer HG, Puga A. Regulation of gene expression by reactive oxygen. Annu Rev Pharmacol Toxicol. 1999;39:67–101. doi: 10.1146/annurev.pharmtox.39.1.67. [DOI] [PubMed] [Google Scholar]
- 37.Schieven GL, Kirihara JM, Myers DE, Ledbetter JA, Uckun FM. Reactive oxygen intermediates activate NF-κB in a tyrosine kinase dependent mechanism and in combination with vanadate activate the p56 and p59 tyrosine kinases in human lymphocytes. Blood. 1993;82:1212–20. [PubMed] [Google Scholar]
- 38.Finco TA, Beg A, Baldwin A. Inducible phosphorylation of IκBα is not sufficient for its dissociation from NF-κB and is inhibited by protease inhibitors. Proc Natl Acad Sci USA. 1994;91:11884–8. doi: 10.1073/pnas.91.25.11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baeuerle P, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–79. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 40.Finkelstein AE, Roisman FR, Ladizesky MG, Walz DT. Auranofin and lysosomal enzymes. J Rheumatol Suppl. 1982;8:46–53. [PubMed] [Google Scholar]
- 41.Hafström I, Seligmann BE, Friedman MM, Gallin J. Auranofin affects early events in human polymorphonuclear neutrophil activation by receptor-mediated stimuli. J Immunol. 1984;132:2007–13. [PubMed] [Google Scholar]