

Abstract
Paramecium bursaria Chlorella virus PBCV-1 mRNA guanylyl transferase (capping enzyme) has been complexed with an mRNA cap analogue G[5′]ppp[5′]G and crystallized. The crystals belong to space group C2221 with unit cell dimensions a = 78.4 Å, b = 164.1 Å, c = 103.3 Å, and diffraction data to 3.1 Å has been collected by using synchrotron radiation. The structure has been solved by molecular replacement by using each of the two domains in the previously determined structure of the enzyme in complex with GTP. The conformation is open with respect to the active site cleft, and all contacts between enzyme and ligand are mediated by domain 1. One of the guanine bases is bound in the same pocket that is utilized by GTP. The conformation of the ligand positions the β phosphate and the active site lysine on opposite sides of the α phosphate. This geometry is optimal for nucleophilic substitution reactions and has previously been found for GTP in the closed conformational form of the capping enzyme, where the lysine can be guanylylated upon treatment with excess manganese(II) ions. The remainder of the cap analogue runs along the conserved active site Lys82 Thr83 Asp84 Gly85 Ile86 Arg87 motif, and the second guanine, corresponding to the 5′ RNA base, is stacked against the hydrophobic Ile86. The ligand displays approximate 2-fold symmetry with intramolecular hydrogen bonding between the 2′ and 3′ hydroxyls of the two ribose rings.
Keywords: capping enzyme, conformation, domain, crystallography
The lifespan of eukaryotic mRNA is believed to be controlled by the so-called “cap” at the 5′ end and by a poly(A) tail at the 3′ end (1). The cap is a covalently attached, N7-methylated guanosine group that is linked to the mRNA via a 5′–5′ triphosphate group. The mRNA capping process involves three separate enzymatic reactions, which may be catalyzed by the same or different polypeptides, depending on organism:
(i) removal of the 5′ phosphate group of the mRNA by RNA triphosphatase, (ii) addition of GMP to the remaining 5′ diphosphate end by RNA guanylyltransferase, or capping enzyme, and (iii) methylation at N7 of the guanine base by RNA guanine-7-methyltransferase,
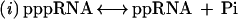 |
 |
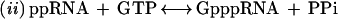 |
 |
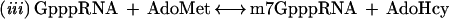 |
 |
The second reaction, which links the GMP to the 5′ diphosphate RNA, consists of several steps, involving an intermediate with the GMP moiety covalently attached to the active site lysine of the capping enzyme through a phosphoramidate bond (2):
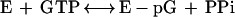 |
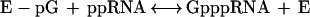 |
Capping enzymes, eukaryotic ATP-dependent DNA ligases, and tRNA ligases appear to be related evolutionarily, as evidenced by the presence of six conserved small sequence motifs (3). The first of these motifs, KXDG, which contains the active site lysine, is also found in prokaryotic NAD+-dependent DNA ligases. The three-dimensional structure of T7 DNA ligase (4) revealed a two-domain arrangement where the first five sequence motifs on domain 1 were all interacting with the ATP substrate. The last motif, on the smaller domain 2, was separated from the active site by a large groove.
The genome of PBCV-1, a virus that affects the green alga Paramecium bursaria Chlorella, contains a relatively small, 330-aa, monofunctional capping enzyme (5) that has been expressed, purified, and characterized (6). Crystallization (7) and structural analysis (8) of Chlorella virus PBCV-1 capping enzyme demonstrated that the bulk of domain 1 indeed has a fold similar to T7 DNA ligase (4). Interestingly, there were two capping enzyme molecules in the asymmetric unit that adopted two distinct conformations, although both contained a noncovalently bound GTP molecule. These conformations, referred to as “open” and “closed,” differ through a rigid-body movement of one domain relative to the other. In both forms, the bulk of the GTP ligand was positioned similarly with respect to motifs 1–5 as the ATP ligand in ligase. However, in the closed form, motif 6 on domain 2 was in contact with the β and γ phosphate of the GTP ligand through a number of hydrogen bonds and salt bridges. The active site lysine in the closed but not in the open form was guanylylated upon soaking the crystals in excess manganese(II) ions. This was explained by the different conformations of the GTP phosphates in the two molecules, where only in the closed form the conformation of the triphosphate tail was suitable for in-line attack on the α phosphate by the active site lysine. On the other hand, only the open form seemed wide enough to accommodate RNA. Because the second reaction (transfer of GMP from the enzyme to RNA diphosphate) is analogous, albeit reversed, to the first (transfer of GMP from pyrophosphate to the enzyme), we postulated that the phosphate conformation and pentavalent intermediate would be similar in the two cases. To study this mechanism further, we have cocrystallized the capping enzyme with a cap analogue, GpppG. This ligand can be regarded as a capped single-residue RNA molecule.
MATERIALS AND METHODS
The capping enzyme was purified as described (7), except that gel filtration was done in the absence of EDTA (ethylenediaminetetraacetic acid). MgSO4 (10 mM) and potassium pyrophosphate (5 mM) were added to the pooled fractions, which were then dialyzed against 1 liter of 50 mM Tris⋅HCl, pH 7.5/10 mM MgSO4/5 mM potassium pyrophosphate/1 mM DTT at 4°C overnight. This was done to ensure a homogenous nonguanylylated enzyme product. After filtering off the precipitate, the enzyme was concentrated and gel-filtered as usual. NaCl was added to a final concentration of 0.4 M, and the enzyme was again concentrated to 15 mg/ml, snap-frozen in liquid nitrogen, and stored at −80°C. Cap analogue (G[5′]ppp[5′]G) was obtained from Epicentre Technologies and was added to the capping enzyme before crystallization. Part of this capping enzyme solution (1.6 μl) (15 mg/ml enzyme, 1.3 mM GpppG, 50 mM Tris, pH 7.5, 400 mM NaCl, 2 mM EDTA, and 4 mM DTT) was mixed with an equal volume of, and equilibrated against, 50 mM potassium phosphate, pH 6.5, 5–10% PEG 8000, and 2 mM ZnCl2. Crystals usually appeared within 24 h and belong to space group C2221 with unit cell dimensions a = 78.4 Å, b = 164.1 Å, c = 103.3 Å. Data were collected on a single crystal that was flash-frozen in a rayon loop at 100K in equilibration solution with 0.4 M NaCl and 30% glycerol. X-ray data were collected to 3.1 Å resolution on a MAR Research image plate detector (MAR Research, Hamburg) at beam station 7.2 (λ = 1.488 Å) at the Synchrotron Radiation Source (Daresbury, U.K.). Integrated intensities were calculated with the program denzo (9). The structure was solved by using the CCP4 version of amore (10) and the previously published structure of the capping enzyme (8). A unique rotational solution was found for each of the two domains that, when combined in the translational search, yielded an open form of the capping enzyme. Structure refinement with low-resolution data is generally difficult because of the low ratio of observations to parameters. We therefore adopted a more thorough, less biased approach, which helped us to find some corrections that were not obvious from the usual 2Fo-Fc and Fo-Fc maps and which resulted in improved agreement between observed and calculated structure factors. During this strategy, the structure was refined by using xplor (11), and solvent-flattened maps were calculated by the CCP4 program suite (12) and displayed by using the graphics program o (13). The crystallographic free R (14) was monitored by using 8% of the data. First, the two domains and the C-terminal stretch (residues 319–327) were rigid-body refined as separate pieces followed by positional and B-factor refinement after some gross corrections. The ligand was then inserted into this initial model, and five residues were then omitted, the structure was refined, and the position of the omitted residues was checked and adjusted according to the Fo-Fc map. The entire protein was checked in this way, but corrections were applied to the initial model rather than the refined model used for map calculations. The initial, corrected model was refined subsequently in the absence of ligand to produce the Fo-Fc map displayed in Fig. 2. This omit map is unbiased insofar as the coordinates used for phase calculations have never been refined together with the ligand. The ligand was finally inserted to produce the final coordinates after some further refinement. A Ramachandran plot was calculated by using procheck (15). The details of data collection and refinement are summarized in Table 1. Coordinates and diffraction data have been deposited with the Protein Data Bank (16), from which copies are available (accession no. 1CKO). The previously determined capping enzyme structures have the accession numbers 1CKM (GTP complexes) and 1CKN (guanylylated adduct).
Figure 2.

Cap analogue bound to the active site of the capping enzyme. Fo-Fc map is displayed at 3 σ level and was calculated before inclusion of the ligand in the model. Motif 1 residues are displayed in thick lines.
Table 1.
Details of data collection and refinement
| Data statistics | |
| Temperature, K | 100 |
| λ/resolution, Å | 1.488/3.1–20.0 |
| Rsym/redundancy | 0.035/2.9 |
| Completeness, % | 96.8 |
| Space group | C2221 |
| Cell axes, Å | a = 78.4, b = 164.1, c = 10.3 |
| Vm, Å3/Da | 4.4 |
| Refinement statistics | |
| Resolution, Å | 10.0–3.1 (all data) |
| R/Rfree, % | 23.3/31.4 |
| rmsd bond length, Å | 0.015 |
| rmsd angles/dihedrals, ° | 3.2/25.0 |
| Number of atoms protein/ligand | 2,561/51 |
| Bave main chain/side chains/ligand Å2 | 64/81/54 |
| Ramachandran favored/ allowed/disallowed, % |
84.8/15.2/0.0 |
RESULTS
The conformation of the cap analogue capping enzyme complex is open, with a sizable gap between the two domains. The folds of the two domain components are the same as for the previously determined structures of capping enzyme complexed with GTP, and the model includes the same number of residues (11–327). The major deviations are found at the N terminus, the hinge region between the two domains, and at the C-terminal stretch. The rms deviation of the superimposed Cα atoms of residues 12–230 is only 0.4 Å atoms between the cap analogue complex and the open GTP complex, whereas that of the superimposed Cα atoms of residues 240–300 is 0.7 Å. All interactions between enzyme and ligand are mediated by domain 1; there are no hydrogen bonds or other close contacts between the ligand and the small domain. Fig. 1 shows the cap analogue complex superimposed on the previously determined structures of the open and the closed GTP complexes. Compared with the open GTP complex, the upper part of domain 1 is tipped approximately 4 Å toward the viewer. The size of the cleft between the domains is, however, very similar in the two cases, and it is possible to replace the GTP with the cap analogue in this structure without any steric hindrance. Because the ligands are not interacting with the small domain in these open structures, the small conformational difference between them seems to have no impact catalytically and is probably merely the result of crystal packing. In contrast, the closed form of the molecule is sterically incompatible with the cap analogue. The main chain of Arg-297 (motif 6) and the side chains of Asp-244, Arg-295, and Asp-297 would be prohibitively close to one of the purine rings. Two of these residues (Asp-244 and Arg-295) were actually found to interact with the phosphate groups of GTP in the closed GTP capping enzyme complex.
Figure 1.

Comparison between the structure of capping enzyme (thick line) complexed with cap analogue (broken line) and the previously determined structures (thin lines) of GTP complexes in open (a) and closed (b) conformation. Only the main chain and the Lys-82 side chain are shown. (a) The upper part of domain 2 is approximately 4 Å closer to the viewer than in the open GTP complex, but the size of the cleft between the domains is similar. (b) The closed GTP complex conformation is sterically incompatible with the presence of the cap analogue in the active site. In each case, the Cα atoms of residues 11–230 of domain 1 were superimposed.
Fig. 2 gives a more detailed view of the cap analogue and its binding site. As was expected, one of the guanine groups is bound in the same pocket as GTP. The rest of the ligand molecule turns back with the second purine group (analogous to the 5′ residue of RNA) interacting with motif 1. The cap guanine is sandwiched between Phe-146 (motif 3) and Ile-216 (motif 4). Compared with the closed GTP complex, the nucleoside group has rotated somewhat around an axis through the C1′ atom and normal to the guanine ring plane, resulting in maximum displacement of around 1.3 Å. Some of the active site residues have followed this movement, and most of the contacts between guanine and enzyme, such as the important hydrogen bond between Lys-188 and the 6-oxo atom, therefore are preserved. The hydrogen bond between the O2′ of the ribose and Asp-131 (motif 2) is also conserved, but that between Arg-87 (motif 1) and ribose O3′ is broken as a result of these movements. The first two phosphate groups follow roughly the same course as the α and β phosphates of the closed GTP complex. As a result of the domain closure, the γ phosphate of the GTP in the closed complex is encapsulated at the bottom of the cleft, where it interacts with residues from both domains. The cap analogue, conversely, turns around after the second (β) phosphate and returns toward domain 1. The first phosphate group is hydrogen bonded to Lys-82, Lys-234, and Lys-236 (motif 5), and the second is hydrogen bonded to Lys-234 and Arg-106. Arg-106 also contacts the O5′ and O4′ of the RNA ribose. The latter is also interacting with the main chain of motif 1 through a hydrogen bond between O2′ and the peptide nitrogen of Arg-87, and through a long, and presumably weaker, hydrogen bond between O3′ and the main chain carbonyl oxygen of Asp-85. The second purine makes no guanine-specific contacts with the enzyme but is packed against the hydrophobic Ile-86 (motif 1). The hydrophobicity of this area is enhanced further by the surrounding residues Leu-19, Leu-21, and Phe-88. The N7 of the purine is also within hydrogen bonding distance of Asp-105. This is surprising because both groups are hydrogen bond acceptors in their normal ionization state. In addition to the interactions between cap analogue and enzyme, intramolecular interactions also appear to be important in determining the conformation and binding properties of the ligand. The two ribose groups are interacting with each other through hydrogen bonds between the O2′ and O3′ groups, with the positions and orientations of the two nucleoside groups (but not the phosphate groups) related by an approximate 2-fold symmetry.
The way that the cap analogue is bound in the capping enzyme active site is compared with the GTP complexes and the guanylylated enzyme in Fig. 3. As already mentioned, the first two phosphates closest to the cap are in analogous positions to the α and β phosphate of GTP in the closed complex. Consequently, the geometry around the α phosphate is similar, in contrast to the different conformation and longer lysine to phosphate distance that is found in the open GTP complex (Table 2). The geometry found in the cap analogue complex and the closed GTP complex appears to be optimal for reaction with Lys-82, whereas in the open and unreactive GTP complex the α phosphate conformation does not seem to allow for an in-line attack of the lysine.
Figure 3.

Comparison of ligands bound in the active site of PBCV-1 capping enzyme. Closed GTP complex (thin line), open GTP complex (thin, dotted line), and guanylylated adduct (thin, broken line) were superimposed on the cap analogue complex (thick line) by using the Cα atoms of residues 11–230 of domain 1.
Table 2.
Distance between ligand Pα and Lys 82 Nζ reactant atoms and the angle between this vector and the bond between Pα and leaving group
| Structure | Distance, Å | Angle, ° |
|---|---|---|
| GpppG complex | 3.1 | 166 |
| Closed GTP complex | 3.2 | 161 |
| Open GTP complex | 4.0 | 70 |
| Guanylylated intermediate | 1.6 | NA |
An angle of 180° would be most suitable for in-line attack by the nucleophilic lysine on the phosphate.
DISCUSSION
The structure of the complex between capping enzyme and cap analogue, as reported in this paper, shows a much more extensive involvement of motif 1 (Lys82 Thr83 Asp84 Gly85 Ile86 Arg87) than had been anticipated. Whereas previous structures of ligase (4) and capping enzyme (8) have confirmed the role of the active site lysine (Lys-82) and shown that Arg-87 hydrogen bonds to the ribose ring of the nucleotide donor, no other motif 1 residue was found to interact with the substrate. The present complex demonstrates that the entire motif 1 is wrapped around the nucleotide acceptor. There are, however, very few side-chain-specific interactions; the Thr-83 and Asp-84 side chains are directed away from the ligand, as can be seen in Fig. 2. Site-directed mutagenesis in the Saccharomyces cerevisiae capping enzyme has shown that substitution of either of these two residues by alanine results in reduced activity, whereas similar mutations of the Lys, Gly, or Arg residue are inactive and lethal (17, 18). Ile-86 seems to be a crucial component of the active site, because of its hydrophobic interaction with the 5′ terminal base of RNA. This conserved hydrophobic residue is usually an Ile, Leu, or Val in the nucleotidyl transferase superfamily, but to our knowledge there has been no mutagenetic study on this residue.
The path followed by the cap analogue describes a U-turn about the β phosphate with the two nucleoside groups obeying an approximate 2-fold symmetry. As a consequence, the β phosphate and the active site lysine are positioned on opposite sides of the α phosphate. This geometry is optimal for nucleophilic substitution reactions and has been found previously for GTP in the closed conformational form of the capping enzyme. Consistent with this geometry, guanylylation takes place only in the closed GTP complex upon soaking in excess manganese(II) ions and not in the open form (8). Thus, both GMP transfer reactions (from pyrophosphate to enzyme and from enzyme to RNA diphosphate) utilize the same reactive α phosphate geometry. In the first reaction, this is achieved by domain closure and interactions between γ phosphate and the small domain. The second transfer occurs with an open enzyme conformation where the α phosphate geometry is determined largely by covalent links to the 5′ end of the mRNA, which is bound noncovalently to domain 1. Electrophilic activation of ATP through enzyme-specific binding of the γ phosphate has been suggested through model building for tyrosyl-tRNA synthetase (19).
The way that the cap analogue binds to the capping enzyme also has implications for our understanding of the catalytic mechanism of ligase. Interestingly, the hydrophobic Ile/Leu residue in motif 1 also is conserved in the NAD+-dependent prokaryotic DNA ligases (20). Although the product of DNA ligation contains a diphosphate rather than a triphosphate bridge, the same principles are likely to be involved, namely orienting the β phosphate on the opposite side of the α phosphate relative to the active site lysine. To achieve this, the ligand is folded back on domain 1. The chemical similarity between NAD+ and the product of the ligase reaction is also intriguing; perhaps the primordial ligase utilized the same binding site, i.e., the K-X-D-G-I/L motif for nicotinamide (AMP donor) and the 5′ DNA base (AMP acceptor). In eukaryotes, the replacement of NAD+ with ATP made this reaction much less symmetrical, and a more elaborate mechanism involving domain closure and motif 6 was required to tether the ATP triphosphate tail. To date there is no structural information available for an NAD+-dependent ligase.
Several capping enzyme structures have been elucidated, e.g., in open and closed conformations, noncovalent complexes with GTP or cap analogue, as well as the guanylylated enzyme intermediate. These structures and, particularly, comparison between them, have improved our understanding of the nucleotidyl transferase mechanism, but there are still many unanswered questions. The most important of these is the proton chemistry. Prediction of the protonization state of a particular amino acid is not always straightforward, because of the influence of the microenvironment on its dissociation constant, pKa. This is particularly true for the active site of the PBCV-1 capping enzyme, which contains a high number of mostly positively charged but also negatively charged residues that interact with each other. The problem is complicated further by the charged nature of the substrate and metal ion cofactor. The active site lysine itself appears to reside between a positively charged and a negatively charged area, when visualized by grasp (21). It is easy to see that small changes in the active site region could change the electrostatic environment of the active site lysine.
Upon guanylylation of the active site lysine, the number of protons on the lysine and phosphate reactants and the phosphoramidate product differ by two, assuming the lysine is fully protonated. Because the reaction is that of a nucleophilic attack by the electron lone pair of the lysine amino group, one proton has to be shed before bond formation takes place. The problem is that because nucleophilicity and pKa are related, anything that enhances nucleophilicity is also likely to increase the pKa, since both protons and nuclei are positively charged. In serine proteases, a classical example of an enzymatic nucleophilic reaction, nucleophilicity of an existing lone pair on the serine is enhanced by hydrogen bonding and proton transfer to the intermediate via a neighboring histidine. The situation for a lysine is different—at neutral pH there is no free lone pair and its affinity for protons has to be reduced while maintaining or increasing nucleophilicity. One possible mechanism would involve two likely hydrogen acceptors that are close to the active site lysine. One of these is the α phosphate group of the substrate itself, and the second is Asp-213, which is hydrogen-bonded to the Nɛ atom of the lysine. Phosphate as a proton acceptor has already been suggested for the related p21ras catalyzed hydrolysis of GTP (22, 23), and carboxylate groups are common acid base catalysts in enzymes. The pKa of 7.6 of a nucleotide triphosphate in solution (24) is reasonable, although this will be affected by magnesium ion ligation and the active site environment. An attractive feature of this proposal, illustrated in Fig. 4, is that it will also enhance the electrophilicity of the reactive phosphorous atom, although transfer of the proton from the lysine to the phosphate will result in the loss of an ion pair and thus will not be favored. However transient such a proton transfer might be, both the lysine and the phosphate would be activated at the same time. This proton must be transferred subsequently to the solvent. Once the lysine is deprotonized, the reaction will be facilitated by the hydrogen bond to Asp, which will also help to stabilize the protonized phosphoramidate intermediate, before this second proton is shuffled first to the Asp and subsequently to the solvent. There are many actors in this complicated play, and at present it is impossible to give a definite and clear picture of the proton chemistry involved. Better crystals suitable for neutron diffraction studies or NMR studies would be helpful in this respect.
Figure 4.

A possible mechanism for formation of the GMP adduct. The guanosine moiety of the GTP is represented by a G.
Acknowledgments
We thank Drs. A. Doherty, C. Ho, and S. Shuman (Sloan–Kettering Institute) for helpful discussions and for supplying the capping enzyme expression vector. We also thank Synchrotron Radiation Source (Daresbury) and Professor L. Johnson (Oxford University) for access to data collection facilities. This project was funded by the Wellcome Trust and the Medical Research Council.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (Protein Data Bank ID code 1CKO).
References
- 1.Beelman C A, Stevens A, Caponigro G, La Grandeur T E, Hatfield L, Fortner D M, Parker R. Nature (London) 1996;382:642–646. doi: 10.1038/382642a0. [DOI] [PubMed] [Google Scholar]
- 2.Shuman S, Hurwitz J. Proc Natl Acad Sci USA. 1981;78:187–191. doi: 10.1073/pnas.78.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shuman S, Liu Y, Schwer B. Proc Natl Acad Sci USA. 1994;91:12046–12050. doi: 10.1073/pnas.91.25.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Subramanya H S, Doherty A J, Ashford S R, Wigley D B. Cell. 1996;85:607–614. doi: 10.1016/s0092-8674(00)81260-x. [DOI] [PubMed] [Google Scholar]
- 5.Li Y, Lu Z Q, Burbank P E, Kutish G F, Rock D L, Vanetten J L. Virology. 1995;212:134–150. doi: 10.1006/viro.1995.1462. [DOI] [PubMed] [Google Scholar]
- 6.Ho C K, van Etten J L, Shuman S. J Virol. 1996;70:6658–6664. doi: 10.1128/jvi.70.10.6658-6664.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doherty A J, Håkansson H, Ho C K, Shuman S, Wigley D B. Acta Cryst D. 1996;53:157–163. doi: 10.1107/S090744499700231X. [DOI] [PubMed] [Google Scholar]
- 8.Håkansson K, Doherty A J, Shuman S, Wigley D B. Cell. 1997;89:545–553. doi: 10.1016/s0092-8674(00)80236-6. [DOI] [PubMed] [Google Scholar]
- 9.Otwinowski Z. Data Collection and Processing. Daresbury, England: Science and Engineering Research Council Daresbury Laboratory; 1993. pp. 56–62. [Google Scholar]
- 10.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 11.Brünger A T, Kuriyan J, Karplus M. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 12.CCP4. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 13.Jones T A, Zou J-Y, Cowan S W, Kjeldgaard M. Acta Crystallogr. 1991;A 47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 14.Brunger A T. Nature (London) 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 15.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 16.Bernstein F C, Koetzle T F, Williams G J B, Meyer E G, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 17.Schwer B, Shuman S. Proc Natl Acad Sci USA. 1994;91:4328–4332. doi: 10.1073/pnas.91.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S P, Deng L, Ho C K, Shuman S. Proc Natl Acad Sci USA. 1997;94:9573–9578. doi: 10.1073/pnas.94.18.9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leatherbarrow R J, Fersht A R, Winter G. Proc Natl Acad Sci USA. 1985;82:7840–7844. doi: 10.1073/pnas.82.23.7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thorbjarnardóttir S H, Jónsson Z O, Andrésson Ó, Kristjánsson J K, Eggertsson G, Palsdottir A. Gene. 1995;161:1–6. doi: 10.1016/0378-1119(95)00286-f. [DOI] [PubMed] [Google Scholar]
- 21.Nicholls A, Honig B J. J Comput Chem. 1991;12:435–445. [Google Scholar]
- 22.Schweins T, Langen R, Warshel A. Nat Struct Biol. 1994;1:476–484. doi: 10.1038/nsb0794-476. [DOI] [PubMed] [Google Scholar]
- 23.Schweins T, Geyer M, Scheffzek K, Warshel A, Kalbitzer H R, Wittinghofer A. Nat Struct Biol. 1995;2:36–44. doi: 10.1038/nsb0195-36. [DOI] [PubMed] [Google Scholar]
- 24.Phillips S J, Eisenberg P, George P, Rutman R J. J Biol Chem. 1965;240:4393–4397. [PubMed] [Google Scholar]