

Abstract
Patterns of gene differences among humans contain information about the demographic history of our species. Haploid loci like mitochondrial DNA and the nonrecombining part of the Y chromosome show a pattern indicating expansion from a population of only several thousand during the late middle or early upper Pleistocene. Nuclear short tandem repeat loci also show evidence of this expansion. Both mitochondrial DNA and the Y chromosome coalesce within the last several hundred thousand years, and they cannot provide information about the population before their coalescence. Several nuclear loci are informative about our ancestral population size during nearly the whole Pleistocene. They indicate a small effective size, on the order of 10,000 breeding individuals, throughout this time period. This genetic evidence denies any version of the multiregional model of modern human origins. It implies instead that our ancestors were effectively a separate species for most of the Pleistocene.
When and where did modern humans evolve? This question remains the focus of much scientific controversy. Traditionally, answers were sought in the human fossil record, which tells us that upright bipedal hominids who made stone tools have occupied much of the temperate Old World for 0.5–1.5 million years. The earlier forms of these hominids are usually called Homo erectus, whereas the later forms, with larger brains and more sophisticated tool kits, are called Archaic Homo sapiens. The Neandertals of Europe are the most familiar of these archaics. In Europe they were replaced by modern humans over several millennia about 40,000 years ago. In Indonesia archaics may have persisted until as recently as 25,000 years ago (1).
Although fossils provide unique and invaluable information, they are very limited in quantity and quality. Huge gaps remain in the human fossil record, and it is difficult to assess, for example, whether there was continuity between archaic and modern humans. Genetic data, in contrast, are easy to collect, and they are accumulating rapidly. Ancient demographic events have left imprints that can be detected in present-day gene differences. Our purpose is to write an accessible summary of current genetic research about human population history.
Genetic methods and data are providing fresh perspectives on a long-standing debate about the origins of our species, which, in its simple form, can be summarized as two competing hypotheses. The multiregional hypothesis suggests that modern humans evolved directly from archaic forms in several different locations in the Old World. Gene flow among these populations, combined with natural selection for advantageous genes, maintained genetic homogeneity of the species. Under this hypothesis, our species had hundreds of thousands, perhaps millions, of ancestors for most of the last million years. Without a large population, gene flow among populations distributed widely over the temperate and tropical Old World would have been impossible.
The other hypothesis is called variously the Garden of Eden, the Noah’s Ark, or the single origin model. According to this hypothesis, a specific population ancestral to modern humans underwent demographic expansion and populated those parts of the world occupied by archaics and then beyond into northern parts of Eurasia and eventually the New World. The contribution of archaic populations to the modern gene pool was negligible. The number of our ancestors just before the expansion (“origin”) of modern humans was small, only several thousand breeding adults.
A clear difference between these two hypotheses is the implied size of the past human population. If the size of the human population had been large throughout much of its history, extant genetic variation should be substantial. Conversely, a small human population would result in relatively little genetic variation. Many genetic systems provide reassuringly congruent estimates: all indicate that human genetic variation is relatively low and that the approximate “effective” size (i.e., the number of breeding adults) of humans is on the order of 10,000 (2, 3). Because several thousands or even tens of thousands of humans could not have occupied the whole temperate Old World, genetic data provide strong support for the single origin hypothesis. Archaeological and skeletal evidence also generally support some version of the single origin hypothesis (4, 5).
The genetic relationship between modern humanity and the world population of archaics at, say, a half million years ago is still unspecified. If the small effective size of humans reflects a transient but drastic reduction in size of a large population, and subsequent recovery, then before the reduction, the number of our ancestors was large, and a graph of our population history looks like an hourglass. By contrast, if we are descended from a subpopulation of archaic humans that was effectively a separate species for the last million years or so, then the graph of our population history is a bottleneck, a short bottle and a very long neck.
The hourglass hypothesis posits that there was a contraction in the number of our ancestors at some time during the Pleistocene, perhaps before the last interglacial, but specifies that the small ancestral population that later expanded had been part of a network of gene flow over the whole temperate Old World occupied by archaic humans before the contraction. In other words, if the genes in the small founding population of modern humans were traced backward in time, they would be dispersed over a large part of the Old World in a population of hundreds of thousands to millions, as in the multiregional hypothesis. Humanity’s small apparent effective size is the result of loss of genetic diversity during the contraction.
The long-neck hypothesis, in contrast, posits that the small ancestral population was small during most of the Pleistocene, for the last million years or so, and that genes in this population traced backward in time were restricted to the range of the particular species of archaic humans that were our ancestors. The essential difference in the two hypotheses is the effective size before the constriction in the middle or upper Pleistocene. They have different consequences for the shape of trees of descent of nuclear genes. Below we show that there is no support in current genetic data for the hourglass hypothesis, whereas the long-neck hypothesis finds strong support.
Estimating Human Effective Size
Effective Size and Census Size.
Effective size is the breeding size of an abstract population, and relating effective to census size of human populations is complicated.
The standard model treats a population as a collection of genes that give birth each generation to a Poisson-distributed number of progeny in such a way that the overall number of genes in the population, N, remains constant from one generation to the next. Under this model, many genes become extinct because they have no progeny. If we pick two genes at random from the population, the probability that they had the same parent (i.e., that they coalesce) is just 1/N, so the expected waiting time until they coalesce is N generations. This can serve as a definition of effective size at a single time, or of the long-term effective size if population size and breeding structure do not change. Felsenstein (6) showed that the effective size of human populations is about one-half the census size. This fraction may have been higher before the evolution of our long postreproductive life span.
When effective size changes over time, then long-term effective size is usually closer to the minimum than to the average effective size. In some simple cases, long-term effective size is the harmonic mean of the changing instantaneous effective size, and this may be a useful heuristic.
Population breeding structure can change effective size in significant ways. If a population is subdivided into partially isolated subpopulations, then the effective size is greater than that of a random mating population because the waiting time to coalescence of genes is increased by the time they spend in different subpopulations. At the level of subdivision among human populations today, this effect would be minor, elevating the ratio of effective to census size by 10% or 15%.
If subpopulations frequently go extinct and are replaced by members of a neighboring subpopulation, then effective size is reduced. In the extreme case where there is almost no gene flow among subpopulations and where descendants of a single subpopulation ultimately replace all others, effective size over time can be closer to the size of a single subpopulation than to the size of the whole population. If something like this happened in our evolution (7), the effective size of the founding subpopulation is exactly what we want to estimate. If there were any substantial gene flow among subpopulations during the replacement process, the effective size over time would reflect the size of the whole population rather than that of a single subpopulation.
Estimates from Mean Pairwise Difference and Segregating Sites.
The simplest genetic evidence about our demographic history is from estimates of overall human effective size. There are two standard approaches to estimating effective size. Each does not estimate size per se but the product of size and mutation rate: these two parameters are almost always confounded in population genetic models. An exception is the estimate from human-specific Alu insertions described below.
The familiar way to estimate size uses DNA sequences. The mean time to coalescence of pairs of sequences is N generations, so the total path length between them is 2N generations. With the infinite sites assumption, according to which every mutation occurs at a new nucleotide position, the expected number of differences between two sequences is 2Nu, where u is the mutation rate for the whole sequence, that is, the pernucleotide rate multiplied by the sequence length. This method requires knowledge of the mutation rate. When the infinite sites assumption is violated, it is often necessary to correct this mean pairwise difference estimate for repeated mutations (8, 9).
An alternate method of estimating N from DNA sequences relies on the overall branch length of the genealogical tree of a sample of n genes and on the infinite sites assumption. The genealogy of a sample of n genes can be divided into n − 1 epochs during which there are n, n − 1, n − 2, … , 2 ancestors of the sample in the population. The expected branch length at each epoch, the product of the number of lines present and the duration of the epoch, has a simple form under the constant size hypothesis. The oldest epoch, when there were two genes ancestral to the sample, has expected duration N generations as derived above. During a more recent epoch, when there are j genes ancestral to the sample present in the population, the hazard of coalescence between any pair is just 1/N per generation and there are j(j − 1)/2 ways that pairs can be formed. The total hazard is j(j − 1)/2N for epoch j, so the expected duration of this epoch is 2N/j(j − 1) generations. Adding these and multiplying the expected length of each epoch by j because there are j lines present, the expected total branch length is
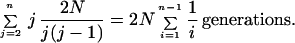 |
1 |
The expected time back to the most recent common ancestor, in contrast to total branch length, is the sum of the interval lengths rather than the sum of the branch lengths. This approaches 2N as the sample size n becomes moderately large. This is called the coalescent of the tree.
It is remarkable that the distribution in Eq. 1 describes both the distribution of when mutations occurred and of the relative frequency of mutations in the sample. The probability that a mutation occurred in epoch j is proportional to 1/j as j varies from 2 to n, and the probability that there are k copies of a mutant in a sample of n genes is proportional to 1/k as k varies from 1 to n − 1.
The expected number of mutations in the whole tree, equivalent to the total number of segregating sites in the sample, is the tree length multiplied by the mutation rate u. Although this estimator of N based on Eq. 1 has in theory better statistical properties than the estimator based on pairwise differences, it is more sensitive to violations of the assumption of constant population size in the past.
An important recent extension of the pairwise method simultaneously estimates the effective sizes of two related species and the effective size of their common ancestral population by using maximum likelihood (3).
New Effective Size Estimates.
While the above methods require knowledge of the mutation rate to estimate N, a different approach is to use the time of separation between the ancestral chimpanzee and human species to calibrate a genetic estimate of human effective size using Alu insertions. Most Alu elements are short (≈300 bp) pseudogenes (10). They are stable, transcriptionally inactive copies of a few active Alu elements, scattered randomly throughout the entire nuclear genome. Collectively, there are about 500,000 copies per haploid genome or 5% of the genome by mass. Some Alu elements are shared with prosimians, monkeys, and apes, whereas others are so recent that they are polymorphic in humans. We have studied insertions of the Ya5 and Yb8 subfamilies whose active elements have been present since before the separation of gorillas from the chimp-human lineage. There are several thousand Ya5 and Yb8 elements in the human nuclear genome, and they are still being inserted. The unique value for evolutionary inference of these loci is that inserted elements are never precisely deleted, so the ancestral state is always known.
Fig. 1 is a schematic of the history of a sample from some locus in our nuclear genome. Interval A is the coalescent of the sample in humans, interval B is the interval from the top of the human coalescent tree to the time of speciation of the ancestral chimp-human population, and interval C is the coalescent time in the ancestral population. Alu insertions in humans that are absent in chimpanzees have occurred somewhere along the branch leading to humans. Any insertions during intervals B and C are fixed in the sample, whereas any that occurred during interval A are polymorphic. The total path length in A is proportional to human effective size, the duration of B can be estimated from paleontology, and C can be estimated by comparing within and between species genetic diversity in chimps and humans at other loci (3).
Figure 1.

Schematic history of a nuclear locus in humans and chimps.
We found 44 fixed and 13 polymorphic insertions in a sample of 122 humans. Sherry et al. (11) give details of the relationship between the number of segregating insertions, the total tree length in interval A, and the implied effective size. We estimated the effective size of humans to be 17,500 with this method, or 9,000 if we assumed the length of interval C to be zero. Our method gives an estimate of human effective size slightly higher than the conventional value of 10,000. The difference might just reflect sampling error or it might indicate a slight reduction in size during the Pleistocene. The Alu method gives greatest weight to effective size near the top of the coalescent tree.
Another new estimate of human effective size during the Pleistocene is derived from diversity among human leukocyte antigen (HLA) alleles. The HLA system is highly polymorphic because of balancing selection that maintains a few allelic lines over very long time periods. The long persistence of alleles at HLA loci implies that the effective size of human ancestors over the latter part of the Cenozoic, i.e., over tens of millions of years, must have been on the order of 100,000 rather than 10,000 (12). Diversity of synonymous (hence neutral) substitutions in selected HLA exons is compatible with the long persistence of allelic lines, whereas that of unselected neutral regions is much lower. This difference led Takahata and Satta (13) to conclude that human ancestral effective size decreased about 1 or 2 million years ago from 100,000 to 10,000. An upper limit on the time of this population reduction which, the authors suggest, may be associated with the dispersal of Homo erectus, is given by the extent of synonymous diversity within HLA lineages.
Changing Effective Size
There are almost 6 billion members of our species on earth today, but the genetic indications of our low effective size suggest that we have not been so numerous for long. Effective size estimates from genetic data refer to a kind of average of population size from the present back to the coalescent of the sample. To understand how genetic data are used to read population size history, it is necessary to look at trees of descent of genes and how population size change affects their characteristics. Because common practice in the literature about human evolution is to reconstruct a gene tree from differences among sequences and then infer history from the reconstructed tree, we include some comments and caveats about this practice.
Properties of Gene Trees.
In this section we show simulated gene trees from populations that have been stationary, that have undergone expansion, and that have undergone contraction. Each of these histories can generate characteristic signatures in the structure of gene trees. In these simulations there are two populations that have always exchanged members at the rate of 0.5 gene per generation; the two populations are approximately as different from each other as two human populations from different continents. The sample is 10 sequences or genes from each population, the red and the green populations.
In practice, trees such as those we portray are reconstructed from gene differences. Each sample, that is, each tip of the tree, is a DNA sequence, and differences among sequences reflect mutations along the branches. The number of mutations along any branch is a Poisson random variable with expectation proportional to the length of the branch. The usual model, the infinite sites model, postulates that every mutation occurs at a different site. In practice in important cases like that of human mitochondrial DNA (mtDNA) there have been multiple mutations at certain sites. These violations of the infinite sites assumption have led to difficulties reconstructing the human mtDNA tree. There is a rich literature on methods for tree reconstruction, and in the case of human mtDNA, there is disagreement about whether current reconstructions are “good enough.” Here we assume that a correct tree has been reconstructed from genetic data and consider inferences that can be drawn from the reconstructed tree. The trees in each of the figures are four equally likely results of the evolutionary process in the population.
Constant Population Size.
The trees shown in Fig. 2 are generated by assuming that the two populations have never changed size. The four trees in the figure are derived from exactly the same demographic scenario, yet they vary widely from one to another. Typically we would have only a single tree to analyze, for example, a tree of mtDNA sequences, so it is important to look at variability among trees in these simulated populations to assess how reliable inference from a single reconstructed intraspecific tree can be.
Figure 2.

Simulated gene trees from a pair of populations of constant size that exchange an average of one-half a gene every generation. These are four simulated loci from the same populations with the vertical axes drawn to the same time scale.
Fig. 2 suggests several cautions about the value of tree reconstructions. First, the time of the root of the tree, the coalescent, varies a lot. Even if we could infer with precision the age of the root from data, it is apparent that any single locus is not very informative about the population. The attention that has been given to the age of the human mitochondrial coalescent seems misplaced because it is only a single locus.
Second, these simulated trees have clear structure. The bottom right tree, for example, has two deep clades. In the top right tree the primary branch on the right contains only samples from the red population, whereas the other contains members of both populations. We could conclude from this that the “origin” of the populations is the red continent from which emigrants populated the green continent. This is just the argument from the human mtDNA tree for an African origin of modern humans: several (contentious) reconstructed trees of descent of human mtDNA contained Africans on one side of the root and people from all the continents on the other (14, 15).
Third, the number of “major clades” varies from tree to tree. The top left and bottom right trees each have two major clades, the lower left tree has four, and the tree on the top right has three. Depending on which tree we had sampled, we might conclude that there were two “founding lineages,” or three, or four. With more study we could reconstruct ancient migration events between the two populations.
Each of these four trees tells a detailed story, and all the stories are utterly spurious and wrong. None of these trees suggests the true population structure, two partially isolated populations that have no other dynamics nor history. The failures are not the result of our small sample size of 20 genes. The overall structure of gene trees is dominated by events at the top of the tree, in the far past. Increasing the sample of genes generally fills in detail at the bottom of the tree. We do not mean to suggest that there is no value in reconstructing intraspecific trees. In some cases, for example, genes of medical interest, the history of the gene rather than the history of the population is of interest. However, this exercise does suggest that population interpretations of single locus trees should be regarded with caution.
Population Expansion.
As we follow a sample going backwards in time to the coalescent, the rate at which lineages coalesce is proportional to the inverse of the effective size. If a population is large now, but expanded rapidly from a small population in the past, then coalescent events will be relatively few since the expansion. They will be concentrated just before the expansion when the population was small. The result is a characteristic star-like gene genealogy as shown in Fig. 3. Prolonged exponential population growth generates similar trees (16). Trees like these are also produced by “selective sweeps,” replacements by an advantageous new allele that is fixed by selection. In the case of population expansion and of a selective sweep, today’s genes have a smaller-than-expected number of ancestors at some time in the past.
Figure 3.

Simulated gene trees from a pair of populations that expanded by a factor of 1,000. The populations exchange an average of one-half a gene every generation.
Population Contraction.
Reduction in population size can lead to rapid loss of genetic diversity. Because the expected depth of a coalescent tree is 2N generations, a contraction in population size lasting this long would erase preexisting diversity in many loci, whereas others would retain two or a few variants that would differ according to the coalescence time, hence the population size, before the contraction. Fig. 4 shows trees from a population that has undergone an instantaneous hundredfold size reduction. The loci in the two right panels retain several variants from before the contraction, whereas those in the left panels lost all precontraction diversity. The visible result of a contraction should be many loci with very little diversity, like those on the left, along with others with a few very divergent gene lineages, like those on the right.
Figure 4.

Simulated gene trees from a pair of populations that have contracted by a factor of 100. The populations exchange an average of one-half a gene every generation. Because the vertical axes are drawn to the same scale, the trees in the left two panels are so shallow that they are invisible.
Just as population expansion mimics selection for a favorable new mutant that evolves rapidly to fixation, population contraction mimics balancing selection maintaining several alleles or classes of alleles over a long time. In both these cases the result is a few alleles or allele classes that coalesce in the far distant past.
Graphical Methods, Trees, and Human History
Expansion in the Pleistocene.
An important paper by Felsenstein (17) showed that estimates of population size could be dramatically improved if the branching order of the underlying tree were known. Because unambiguous reconstruction of this branching order is often impossible, computer-intensive methods have been developed that examine large numbers of trees, evaluating the likelihood of each tree and the likelihood of a demographic hypothesis given the tree (18, 19). As these methods become faster and more widely available, methods like those as we describe here will be relegated to screening roles. However, the simple approaches we describe are fast, simple, and easy to understand. Computer-intensive methods have the important disadvantage that one never really knows what they do; it is difficult to prove that they are working correctly.
The top panel of Fig. 5 shows a simulated tree of a population that has undergone an expansion. The middle and bottom panels show two summaries of the simulated tree that are simple to compute. The middle panel shows the frequency spectrum of mutations. The spectrum is the distribution of segregating sites according to frequency in the sample. In practice we usually do not know the ancestral state at a site, so we do not know which of two variants is the mutant. The spectrum then must be “folded” at one-half the sample size. In the present case a mutant that occurred twice in the sample of 20 would be indistinguishable from a mutant that occurred in 18 of the 20, so these two categories are combined and the range of the spectrum is from 1 to 10.
Figure 5.

Gene tree (Top), frequency spectrum (Middle) and mismatch distribution (Bottom) from a population that has undergone a population expansion. The circles on tree represent mutations. The simulation parameters match approximately those estimated from human mtDNA.
Mutations that happened far in the past, near the top of the gene tree, can occur at high frequencies in the sample. Recent mutations, on the other hand, always exist in one or a few copies. If an expansion has occurred, so that the gene tree is star-like as it is here, many mutations occur in the long recent branches and there are more singleton sites and sites with low frequency variants than are expected in a stationary population. Fig. 6 shows a simulated tree from a stationary population, and the spectrum in the middle panel of this figure shows that there are more variants at intermediate frequencies.
Figure 6.

Gene tree (Top), frequency spectrum (Middle) and mismatch distribution (Bottom) from a population that has always been constant in size.
The bottom panels of Figs. 5 and 6 show mismatch distributions calculated from the simulated trees. These are histograms of the numbers of sequence differences among all possible pairs of sequences in the sample. Under the infinite sites assumption, every mutation on the path from one sample to another contributes a single difference to the comparison. These are not ordinary distributions because the data points are not independent, but they do provide quick visual summaries of important properties of the trees. In the case of population expansion in Fig. 5 the biggest contribution to pairwise differences is from the long terminal branches that are independent of each other, so the result is a smooth and often unimodal mismatch distribution in the bottom panel of the figure. Mismatch distributions from stationary populations are reliably ragged and often multimodal, as in the bottom panel of Fig. 6.
Fig. 7 shows these distributions from a worldwide sample of 636 sequences at 411 positions of the first hypervariable segment of mtDNA (ref. 20; L.B.J., unpublished data). In the top panel, the spectrum of frequencies is collapsed into four ranges. Expected values shown are those expected if the population had always been the same size. In the human data there is a large excess of low frequency variants in accordance with the hypothesis that the human population has undergone a major expansion. The bottom panel of the figure shows the mismatch distribution: it is smooth with the distinct mode characteristic of a population expansion or a selective sweep within the last several hundred thousand years. Almost all mismatch distributions from human mtDNA have the general appearance of Fig. 7. We and others interpreted this pattern as a signature of a population expansion of our ancestors beginning in the last interglacial about 100,000 years ago (22–24).
Figure 7.

Frequency spectrum and mismatch distribution from a world sample of 636 mtDNA sequences at 411 positions of the first hypervariable segment (HVS-I). Compare this with Fig. 5. The diamonds show the expected number of segregating sites in each frequency interval expected in a constant size population.
However, there is the possibility that the pattern results from a selective sweep in which an advantageous new mtDNA sequence replaced all other sequences in the population. To test this we require information from other loci, and such information is only recently becoming available.
The first new evidence is from a report of sequence differences in approximately 20,000 sites along the nonrecombining part of the Y chromosome from 718 men (25). The transmission of this part of the Y is formally like that of mtDNA except that it is through males rather than through females. There are, however, two important ways in which these data must be treated differently from mtDNA sequence data. First, the segregating sites were ascertained in small numbers of Y chromosomes, 21 in one set and 53 in the other. Unfortunately correct mismatch distributions cannot be computed from the data because the typings in the screening samples were not reported. Many of the 718 chromosomes are expected to differ at undetected sites.
Second, the gene tree was reconstructed without ambiguity and the ancestral states determined by comparisons with the same sites in African apes, so we can examine the whole frequency spectrum of the segregating sites without folding. Fig. 8 shows the observed distribution of sites among four frequency classes. The diamonds in the figure show the expected distribution under the hypothesis of constant population size. There is a large excess of low frequency sites, consistent with the hypothesis of population expansion and inconsistent with the hypothesis that the evidence for expansion in human mtDNA reflects a selective sweep.
Figure 8.

Frequency spectrum and mismatch distribution from a world sample of 718 Y chromosome sequences. There are 20 segregating sites ascertained at approximately 20,000 positions. Ascertainment was done in two samples, one with 21 chromosomes and one with 53. If a site has population frequency x, then the probability that it will be detected in a sample of size n, the ascertainment function, is 1 − (xn + (1 − x)n). Diamonds show the expected number of sites in each frequency class under the hypothesis of constant population size, computed by multiplying the ascertainment function by the distribution in Eq. 1. The excess of low frequency sites is consistent with a Pleistocene population expansion. Because the whole nonrecombining portion of the Y chromosome is a single locus, these sites are not independent, and there is no simple statistical test of the constant size hypothesis.
The second new evidence supporting ancient population expansion is from tandem repeat loci. These are loci in which some short motif is repeated, and the number of repeats varies from chromosome to chromosome. Mutation occurs at these loci, according to the usual model, by small gains and losses in the number of repeats. Kimmel et al. (26) show that the variance of repeat size and homozygosity change at different rates after population expansion. Comparison of these quantities at 60 tetranucleotide loci from three human continental groups showed clear evidence of population expansion among Europeans and Asians, but none among Africans. Patterns in mtDNA and in human craniometric traits (27) suggest that the ancestors of Africans expanded before the ancestors of other continental populations. Because repeat loci evolve rapidly, their findings may suggest that these loci in Africans have had time to reach their new equilibrium, erasing the trace of the expansion.
In summary, the estimates of overall human effective size of 10,000 from nuclear sequences, Alu insertions, and HLA exons, mtDNA mismatch distributions, frequency spectra from mtDNA and from the Y chromosome, and discordance between allele size variance and homozygosity at tandem repeat loci all support the hypothesis of a bottleneck in our past during which the number of our ancestors was only a few thousand breeding adults. The original formulation of the multiregional hypothesis, that there was a worldwide transformation of archaics into modern humans caused by spread of new alleles, is contradicted by all these findings.
The best available estimates of mtDNA mutation rates imply that the expansion occurred between 100,000 and 50,000 years ago in excellent agreement with archaeological evidence of the earliest modern humans about 100,000 years ago, and the “creative explosion” of upper Paleolithic type technology about 50,000 years ago (28). This could be illusory because the time depends on estimates of mtDNA rates, and these are fragile at best. A recent review (29) suggests that the expansion apparent from genetics is associated with the first complex flake tool industries several hundred thousand years ago, i.e., much earlier than the Upper Paleolithic industries associated with modern humans in Europe. The older date cannot be falsified from the genetic evidence.
Population Size Before the Bottleneck.
Both mtDNA and Y chromosomes coalesce several hundred thousand years ago, so they provide no information about population size before then. The coalescent of nuclear genes should be four times as old as that of mtDNA and the Y in the absence of population size change. Unfortunately, nuclear genes undergo recombination, and recombination rapidly destroys evidence of population size in DNA sequences. The mutation rate at nuclear loci is also much lower than that of mtDNA so long sequences are necessary to achieve resolution comparable to that of mtDNA sequences, but the longer the sequence the more likely recombination has occurred.
If suitable nonrecombinant nuclear sequences can be found, then it will be possible to test the hourglass model by looking for very deep differences between alleles (see Fig. 4). Such patterns are conspicuously absent in humans with the exception of the HLA system, which owes its deep allelic lineages instead to balancing selection.
Harding et al. (30) describe a careful analysis of nuclear sequences from a region where recombination has not erased the coalescent history. They studied part of the β-globin gene using a computer-intensive method that examined large numbers of possible mutation histories. Approximately 10% of their sequences were discarded from the analysis because they had undergone recombination. There was no evidence of deep roots as predicted by the hourglass model; instead, they suggest that there was a constant population size of 10,000 all the way back to the root of this nuclear tree.
Under the hourglass model, nuclear loci with deep branches at the top of the gene tree should often lead to disjoint bimodal or multimodal distributions of allele size at tandem repeat loci. Disjoint distributions should occasionally appear even with constant population size. For example, the gene trees in the right two panels of Fig. 2 could generate bimodal allele size distributions because of size-change mutations accumulating along the deep top branches. The gene trees in the left two panels probably would not. Many tandem repeat loci do show such allele size distributions, but their frequency does not appear to be any greater than that expected under constant population size.
The most useful class of genetic markers for ancient population studies is currently young Alu insertions. Because these are inserted into the genome but never precisely deleted, the ancestral type is always known. They are inserted at random into the nuclear genome so that the probability of two insertions in the same place is vanishingly small. Fig. 9 shows the spectrum of frequencies of 23 human-specific Alu insertions (refs. 11 and 21; M.A.B., unpublished data) along with the predicted spectra under the long-neck and hourglass models of Pleistocene human population size. There is no suggestion of population contraction in our history from these data. Because these insertions have independent histories after they are inserted, standard statistical methods for contingency tables can be applied to them. The long-neck model cannot be rejected, whereas the extreme hourglass model can be rejected.
Figure 9.

Frequency spectrum of 23 Alu insertions in humans. The diamonds show the expected numbers of loci under constant population size as specified by the long-neck model. Circles show expected numbers of loci under the hourglass model of a population contraction. The hourglass hypothesis can be rejected by a statistical test, whereas the long-neck model cannot. Each of these was ascertained in a diploid. The probability of detecting at least one copy of an insertion in a diploid whose population frequency is x is x2 + 2x(1 − x), the ascertainment function for this system. Expected values for the long-neck model were computed by multiplying the distribution in Eq. 2 by this function. Expected values for the hourglass model were computed by multiplying the ascertainment function by the uniform distribution because the distribution of the number of copies of a mutation in the top interval of the tree is uniform.
Conclusions
We have avoided hedging and qualifying our findings in the interest of making this paper simple and accessible. Nevertheless, the broad picture that we paint continues to gain empirical support. Most of the familiar specimens of Homo erectus and of archaic humans known from the Pleistocene were not members of populations ancestral to us, instead “the fate of most such populations appears to be tragic” (13). We are descended from a population that was effectively a separate species for at least the last 1 or 2 million years. Although the size of this population must have fluctuated over time, it was often reduced to the level of several thousands of adults. Such a population would have occupied an area the size of Swaziland or Rhode Island rather than a whole continent, although episodic expansions would have covered a much larger area. Archaeologists should find and identify this population.
Acknowledgments
We are grateful for help and suggestions from Elise Eller, Marta Lahr, Renee Pennington, James O’Connell, John Relethford, Naoyuki Takahata, Stephen Wooding, and the Human Diversity Project, King’s College Research Centre, Cambridge.
ABBREVIATIONS
- mtDNA
mitochondrial DNA
- HLA
human leukocyte antigen
References
- 1.Swisher C C, Rink W J, Anton S C, Schwarcz H P, Curtis G H, Suprijo A, Widiasmora Science. 1996;274:1870–1874. doi: 10.1126/science.274.5294.1870. [DOI] [PubMed] [Google Scholar]
- 2.Nei M, Graur D. In: Evolutionary Biology. Hecht M K, Wallace B, Prance G T, editors. Vol. 17. New York: Plenum; 1984. pp. 73–118. [Google Scholar]
- 3.Takahata N, Satta Y. Proc Natl Acad Sci USA. 1997;94:4811–4815. doi: 10.1073/pnas.94.9.4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein R G. J World Prehist. 1995;9:167–198. [Google Scholar]
- 5.Lahr M M. The Evolution of Modern Human Diversity. Cambridge, U.K.: Cambridge Univ. Press; 1996. [Google Scholar]
- 6.Felsenstein J. Genetics. 1971;68:581–597. doi: 10.1093/genetics/68.4.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahata N. Mol Biol Evol. 1994;11:803–805. doi: 10.1093/oxfordjournals.molbev.a040160. [DOI] [PubMed] [Google Scholar]
- 8.Watterson G A. Theor Pop Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 9.Tajima F. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deininger P L, Batzer M A. In: Evolutionary Biology. Hecht M K, MacIntyre R J, Clegg M T, editors. Vol. 27. New York: Plenum; 1993. pp. 157–196. [Google Scholar]
- 11.Sherry S T, Harpending H C, Batzer M A, Stoneking M. Genetics. 1997;147:1977–1982. doi: 10.1093/genetics/147.4.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahata N. Mol Biol Evol. 1993;10:2–22. doi: 10.1093/oxfordjournals.molbev.a039995. [DOI] [PubMed] [Google Scholar]
- 13.Takahata, N. & Satta, Y. (1998) Immunogenetics, in press.
- 14.Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson A C. Science. 1991;253:1503–1507. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- 15.Cann R L, Stoneking M, Wilson A C. Nature (London) 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- 16.Hudson R R, Slatkin M. Genetics. 1991;129:555–562. doi: 10.1093/genetics/129.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felsenstein J. Genet Res. 1992;59:139–147. doi: 10.1017/s0016672300030354. [DOI] [PubMed] [Google Scholar]
- 18.Kuhner M K, Yamato J, Felsenstein J. In: Progress in Population Genetics and Human Evolution. Donnelly P J, Tavaré S, editors. New York: Springer; 1997. pp. 183–192. [Google Scholar]
- 19.Griffiths R C, Tavaré S. In: Progress in Population Genetics and Human Evolution. Donnelly P J, Tavaré S, editors. New York: Springer; 1997. pp. 165–182. [Google Scholar]
- 20.Jorde L B, Bamshad M J, Watkins W S, Zenger R, Fraley A E, Krakowiak P A, Carpenter K D, Soodyall H, Jenkins T, Rogers A R. Am J Hum Genet. 1995;57:523–538. doi: 10.1002/ajmg.1320570340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoneking M, Fontius J J, Clifford S L, Soodyall H, Arcot S S, Saha N, Jenkins T, Tahir M A, Deininger P L, Batzer M A. Genome Res. 1997;7:1061–1071. doi: 10.1101/gr.7.11.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sherry S, Rogers A R, Harpending H C, Soodyall H, Jenkins T, Stoneking M. Hum Biol. 1994;66:761–775. [PubMed] [Google Scholar]
- 23.Harpending H C, Sherry S T, Rogers A R, Stoneking M. Curr Anthropol. 1993;34:483–496. [Google Scholar]
- 24.Rogers A R, Harpending H C. Mol Biol Evol. 1992;9:552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- 25.Underhill P A, Jin L, Lin A A, Mehdi S Q, Jenkins T, Vollrath D, Davis R W, Cavalli-Sforza L L, Oefner P J. Genome Res. 1997;7:996–1005. doi: 10.1101/gr.7.10.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimmel, M., Chakraborty, R., King, J. P., Bamshad, M., Watkins, W. S. & Jorde, L. B. (1998) Genetics, in press. [DOI] [PMC free article] [PubMed]
- 27.Relethford J H, Harpending H. Am J Phys Anthropol. 1994;95:249–270. doi: 10.1002/ajpa.1330950302. [DOI] [PubMed] [Google Scholar]
- 28.Klein R G. The Human Career. Chicago: University of Chicago Press; 1989. [Google Scholar]
- 29.Foley R A, Lahr M M. Camb Arch J. 1997;7:3–32. [Google Scholar]
- 30.Harding R M, Fullerton S M, Griffiths R C, Bond J, Cox M J, Schneider J A, Moulin D S, Clegg J B. Am J Hum Genet. 1997;60:722–789. [PMC free article] [PubMed] [Google Scholar]