
Figure 2.
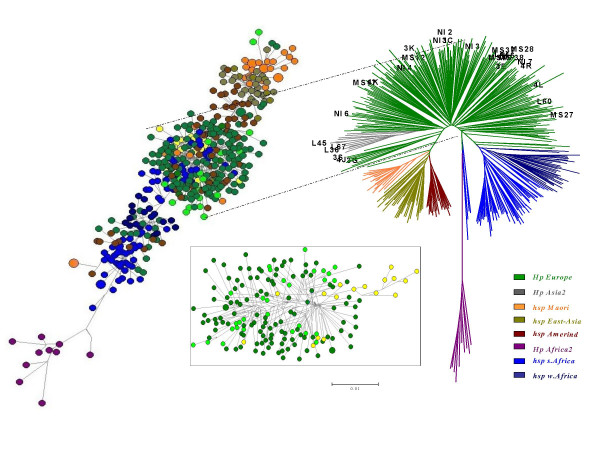
Neighbor joining tree (Kimura 2-parameter) (right) showing the global population structure of H. pylori wherein Indian isolates are highlighted. The phylogenetic tree was based on a total of 23 sequence records of South and North Indian isolates while incorporating ~400 other sequence records from pubMLST database representing different H. pylori populations and sub populations in the world. The population genetic structure was investigated by determining the multilocus haplotypes based on concatenated sequences of seven unlinked housekeeping genes that are scattered around the H. pylori chromosome. Individual isolates were assigned to bacterial populations called hpEastAsia (sub-populations: hspEAsia, hspMaori, hspAmerind), hpEurope, hpAfrica1 (hspSAfrica, hspWAfrica), hpAsia2 and hpAfrica2 [11]. Representatives from each of these (sub)-populations were chosen for subsequent analysis of the cagPAI. Isolates from the population hpAfrica2 do not contain cagPAI. Phylogenetic relationships were also estimated through NETWORK analysis (left) based on 665 mutating positions that revealed the co-evolution of the H. pylori genome. The Ladakhi (yellow) and other Indian (light green) lineages were more clearly discerned within the European (dark green) cluster (centre box), when analyses based on the remaining 650 mutating positions were performed. For the Neighbor-joining tree (right), the bootstrap values of the interior branches as calculated in MEGA, were significantly high to indicate the correct topology of the branches within the clades.