

Abstract
Flow cytometry (FC) has been introduced to characterize and to assess the physiological states of microorganisms in conjunction with the classical plate-counting method. To show the applicability of the technique, in particular for the development of kinetic models, pure culture fermentation experiments were followed over time, using both prokaryotic (Lactobacillus hilgardii) and eukaryotic (Saccharomyces cerevisiae) microorganisms growing in standard culture media (MRS and YPD). The differences observed between the active and viable cells determined by FC and CFU, respectively, allowed us to determine that a large number of cells were in a viable but nonculturable (VBNC) state, which resulted in a subpopulation much larger than the damaged-cell (double-stained) subpopulation. Finally, the determination of the evolution of viable, the VBNC, and the dead cells allowed us to develop a segregated kinetic model to describe the yeast and the bacteria population dynamics and glucose consumption in batch cultures. This model, more complete than that which is traditionally used, based only on viable cell measurements, describes better the behavior and the functionality of the cultures, giving a deeper knowledge in real time about the status and the course of the bioprocesses.
Accurate measurements of biomass, substrate consumption, and product formation are the most important parameters for bioprocess control and evaluation. Measurement of active biomass is essential for determining growth rates and cell yields and for providing a base for kinetic model formulations. The presence of a high proportion of dormant, dying, or dead cells at any time during a fermentation process will have an obvious detrimental effect on the synthesis of any desired product (56).
The most common method for measuring cell mass is dry weight, but it is not sufficiently accurate for analyzing very dilute concentrations of cells, requiring high volumes, and cannot differentiate subpopulations or discriminate microorganisms from debris (51). The measurement of wet weight is a faster method, but it is considered less accurate than measuring dry weight, thus, it is not used. Turbidity measured as optical density is one of the few on-line methods available for monitoring cell growth during a process. It rarely takes into account background medium composition being replaced by dry weight values for the final evaluation of the results (21) after the construction of a standard calibration curve. For cell number measurements, cell counting on agar plates is the method most commonly used to measure viability of a cell population, considering that a cell is viable when it shows reproductive ability, thus imposing a two-value logic basis (41). This is an off-line, indirect, and slow method, and it fails because sometimes the cells cannot reproduce in an artificial environment (11, 36). The inability of a cell to divide is not the same as cell death, as this argument is subject to extensive discussions and investigations (9, 26, 36, 39, 40, 41, 56).
Classical techniques (turbidity measurement, dry weight determination, and plate counting method) give information about microbial growth associated with cell division but do not consider physiological states. Mathematical models based on these types of techniques become inaccurate when it is assumed that microbial populations are homogeneous with respect to their physiological state but heterogeneities are known to exist (9, 19, 30, 36, 41). Cell cultures normally are heterogeneous due to factors such as the cell cycle, inhomogeneous cell microenvironments, and genetic differences (23). The development of kinetic models requires rigorous and accurate information about physiological states in a certain population. Flow cytometry (FC) can reveal information about the dynamics and physiological heterogeneity of microbial populations and describes more accurately the population than the average values obtained from traditional techniques (46, 50, 59).
Flow cytometry is applicable for quantitative analysis of total microbial biomass. Measurements are made very rapidly with a large number of individual cells and give objective and accurate results (7, 12, 54, 63). This technique has been useful for the analysis of nonculturable bacteria cells (14) as well as for monitoring changes in bacterial subpopulations under growth-limiting conditions (5). Other FC studies have been used to describe the Saccharomyces cerevisiae cell cycle (52, 62), metabolic studies (55), cell characterization (47), and single-cell glucose uptake rates (34), as well as biotechnological applications. In this field, assessment of cell functionality has been applied to fermentation technology and on-line bioprocess control, quality control in the food industry, bioremediation, and mammalian cell cultures, among others (36, 50, 53). However, application of data obtained by the FC technique to kinetic modeling is still very scarce. Alcon et al. (1) formulated a growth-structured kinetic model applied to Candida bombicola growth based on intracellular compound measurements by FC.
The first aim of the present study was to evaluate the differences between the application of traditional techniques and FC analysis for microbial kinetics. In order to establish the application of the procedure, yeast and lactic acid bacteria (the main microorganisms used in conducting wine and cider fermentations) were grown as pure cultures under batch conditions in standard semisynthetic media (YPD and MRS) after preservation treatments (the active dried form and freezing, respectively). Experimental data for the total biomass obtained by optical density measurement (OD) and cell counting and by FC analysis during cell growth were fitted to mathematical models, and kinetic parameters were compared. Finally, a segregated kinetic model was proposed for describing the profile of cell subpopulations at different physiological states (viable, viable but not culturable [VBNC], and dead cells) and glucose uptake.
MATERIALS AND METHODS
Microorganisms and culture conditions.
The yeast used in this work was a strain of Saccharomyces cerevisiae var. bayanus in an active dried form (strain Pasteur Institute, Paris, 1969, “Champagne”) supplied by Novo Ferment (Switzerland), generally used in industrial cider production in Asturias (northern coast of Spain). The malolactic bacteria was identified by molecular techniques as Lactobacillus hilgardii (Lc2) and was previously isolated from the cellars of the Asturian cider-making industry Sidra Escanciador, S.A., (Villaviciosa, Principado de Asturias, Spain), and stored at −20°C in glycerol (20% [vol/vol]).
Yeasts were grown in Erlenmeyer flasks (250 ml) containing 50 ml of YPD medium (2% [wt/vol] glucose, 2% [wt/vol] peptone, 1% [wt/vol] yeast extract) under aerobic conditions (250 rpm in an orbital shaker [New Brunswick Scientific, Edison, NJ]) at 28°C for 360 h.
Bacterial cells were grown in MRS medium (Biokar, France) under microaerophilic conditions (without shaking) and incubated at 30°C for 96 h.
Sampling.
Yeast and bacterial growth was followed by measuring OD at 660 nm (OD660), by plate counting on solid media, and by FC. Samples were taken throughout growth curves, and cells were harvested by centrifugation and washed twice in phosphate-buffered saline (pH 7.4; 0.22-μm-pore-size filtered). Cell concentration was adjusted to 4 × 105 cells ml−1 in the same buffer used for FC measurements. Flasks containing bacteria were shaken just before sampling in order to homogenize the biomass content. Supernatants were filtered (0.45-μm pore diameter) and frozen (−20°C) until glucose (substrate) analysis.
Growth curves were carried out in duplicates, as two independent experiments, for each microorganism. The data were finally expressed as the means of the values obtained.
Two-sample comparison was performed with statistical software (Statgraphics Plus 3.11).
Turbidity measurements.
The evolution of biomass was determined by following the OD660 of medium with bacterial growth (UV 1203 model spectrophotometer; Shimazdu) versus that of a blank (the same medium without cells). OD data were converted to dry weight using the corresponding calibration curve previously obtained, and the results were finally expressed as g · liter−1.
Plate counting.
Cells were held in the “hot spot” of a sonication bath for 2 to 3 seconds just before plating to avoid aggregates (21). Numbers of viable yeast and bacterial cells were determined by the standard counting method by plating statistically significant dilutions (20 to 200 colonies per plate) in triplicates on YPD and MRS plates (containing 2% [wt/vol] agar), respectively. Colonies on plates were enumerated after 2 and 6 days, respectively, of incubation at 30°C, and viable counts were expressed as CFU/ml, as averaged values. Under these conditions, assuming that a colony originates from a single cell, viable cells were finally expressed in g · liter−1, taking into account the calibration curve obtained for total cells stained by DRAQ5 (cells·ml−1) versus dry weight (g · liter−1).
FC counts versus dry weight.
Cell suspensions at different OD values were filtered through 0.45-μm nitrocellulose membranes previously dried (110°C, 24 h), washed with distilled water (two volumes) and dried until reaching constant weight (110°C, 24 h). Aliquots for the same cell suspensions were stained with DRAQ5. In previous work (20) sonication of malolactic bacterium samples was tested (15 and 30 s) to avoid problems associated with cell aggregation. Results obtained led us to consider than the standard two phosphate-buffered saline washes performed, including vortexing, were enough to avoid aggregation. In this work, to ensure sample homogeneity and in order to use the same procedure as for the plate counting method, cells were sonicated for 2 to 3 seconds just before FC analysis. Corresponding calibration curves were developed. Measurements were carried out in triplicates, and the average values were used. Thus, cells were finally expressed in g · liter−1, in order to use the same units.
Staining for flow cytometry analysis.
Total counts of cells were determined by using a DRAQ5 single-staining method (Biostatus Limited, United Kingdom) to differentiate the microorganisms from other particles in samples. Dual staining (with ChemChrome V6 [CV6] and propidium iodide [PI]) was used for each sample, as these stains differ in their spectral characteristics and their abilities to penetrate cells. CV6 (Chemunex, France) allowed labeling of metabolically active yeast and bacterial cells, thus comprising the fraction of active cells not able to grow on plates (VBNC cells). PI, a fluorescent nucleic acid dye, was used to stain dead cells. Staining solutions were added to 200 μl of the cell suspensions and incubated in the dark. Based on previous work (20) using control samples, regions were gated for the different fluorescence patterns of each microorganism. Finally, in order to validate the CV6-PI dual-staining assay, unstained, DRAQ5-stained, CV6-stained, and PI-stained cells and double-stained controls were tested, and gates were defined as dot plots of green and red fluorescence.
Flow cytometry analysis.
Experiments were carried out using a Cytomics FC 500 model (Beckman Coulter) with 488-nm excitation from an argon-ion laser. All parameters were expressed on a logarithmic scale. DRAQ5 red fluorescence data were collected in the FL4 channel (675 nm). Fluorescence data for cells stained by CV6 and PI were collected in channels FL1 and FL3 (525 nm and 610 nm, respectively) in a dual dot plot. The detection threshold was set at medium rate. Fluorescent microspheres (Perfect Count, Cytognos, Salamanca, Spain) were added to each sample as an internal reference, following the supplier's recommendations for ratiometric counting. In order to get a significant number of cells to ensure the efficiency of the test, 2,000 microspheres were acquired for each analysis. When less than 100 positively stained cells (with PI or CV6) were detected, the result was not considered significant (37). Data analysis was carried out using Cytomics RXP Analysis (Beckman Coulter).
Glucose analysis.
Glucose in MRS and YPD supernatants was analyzed by the dinitrosalicylic acid method for determining reducing sugars (33), modified as reported previously (45).
RESULTS AND DISCUSSION
Population dynamics determined by turbidity, by plate counting, and by flow cytometry.
Cell concentration was followed throughout yeast and bacterial growth curves by turbidity (OD measurements), plate counting, and FC analysis in combination with fluorescent dyes. In this latter case, total cell numbers were determined by DRAQ5 single staining. CV6, a fluorogenic ester that is cleaved in the presence of esterase activity, was used as the indicator of the cell subpopulation considered metabolically active. PI-stained cells presumably have a compromised membrane, allowing the ionic dye to enter the cells. The multiparametric analysis permitted distinction among different populations with dual parameter dot plots: CV6-stained cells (metabolically active), PI-stained cells (dead), and CV6-PI stained cells (dual-stained, intermediate populations considered damaged) (Fig. 1).
FIG. 1.

Green and red dot plots for yeast (a) and bacteria (b). Different regions (subpopulations) were gated in each case according to their staining properties: A, CV6-stained cells (metabolically active); B, PI-stained cells (dead); C, double-stained cells (damaged).
Figures 2 and 3 show the different cell populations measured by FC (total, metabolically active, dead, and damaged double-stained cells), plate counting (viable cells), and OD evolution (expressed as dry weight in g · liter−1) throughout yeast and bacterial growth curves, respectively. As previously mentioned, cell concentration determined by FC was expressed in g · liter−1 using a calibration curve (DRAQ5 staining versus dry weight) calculated for yeast and bacteria.
FIG. 2.

Experimental results for yeast growth over time. Total cells ▴, metabolically active cells ▵, viable cells (•), dead (□), and double-stained (×) cells are expressed in g · liter−1 in the main y axis. Total biomass expressed as dry weight (g · liter−1) (⧫) is shown in the secondary y axis.
FIG. 3.

Experimental results for bacteria growth with time. Total cells (▴), active cells (▵), viable cells (•), dead cells (□), and double-stained (×) cells are expressed in g · liter−1 in the main y axis. Total biomass is expressed as dry weight (g · liter−1) (⧫) in the secondary y axis.
In Fig. 2, yeast biomass (expressed as dry weight) and the number of total cells (obtained by FC) increased until stationary phase was reached (at 63 h). At the beginning of this stage, total biomass values calculated by both techniques were of the same order, 10 and 15 g · liter−1 by OD and FC, respectively. From this point, biomass values were maintained at a constant level all through the stationary phase, although small oscillations were observed, probably due to sample handling. The mechanisms by which yeast cells survive prolonged periods of nutrient limitation and resume proliferation remain unclear (64). The same authors considered that the ability to maintain viability without added nutrients is likely to be the most rigorous physiological definition of entry into stationary phase.
It should be noted that the population of active yeast cells (CV6 stained) as determined by FC was very close to the total cell number (DRAQ5 stained), and there was no evidence of PI-stained or doubled-stained cells in yeast growing at exponential phase (using a commercial active dried form as inoculum). An increase in cell viability and in cells showing metabolic activity was observed until 63 h, although only ∼30 to 34% of metabolically active cells were capable of forming colonies on solid medium. Not only metabolic activity but also yeast culturability were maintained over time until 358 h of incubation (during stationary phase). At this time, 90.4% of total cells were stained with CV6, but only 33.7% were culturable on solid medium. In addition, PI-stained and double-stained cells were 1.9% and 2.2% of the total number, respectively.
For bacterial growth, an increase in total biomass and in cell numbers, determined by OD and FC, was observed during exponential phase and different biomass values were reached at the end of this stage (0.8 and 1.9 g · liter−1 by each technique, respectively) (Fig. 3). After 29 h of bacterial incubation, values remained approximately constant all through stationary phase. A culturable subpopulation was maintained for ∼11 to 21% of active cells during exponential and stationary phases. Only a small subpopulation (4% of total cells) was found by PI staining to show compromised membranes in the exponential phase. Cells stained red exhibited a loss of membrane integrity which resulted in the collapse of the cell energetics and active transport, that is, in the death of the cell (17, 35). Ericsson et al. (17) reported that for Escherichia coli cultures growing at the exponential phase, no cells were found to have lost membrane integrity and false IP positives were not identified. It should be kept in mind that in this work, a freezing culture was used as inoculum. The doubled-stained population of L. hilgardii increased between 0.5 and 5% throughout bacterial growth in the exponential phase, considered a membrane-damaged subpopulation, a transient step between active and membrane-compromised or irreversibly membrane-damaged states. After 63 h of bacterial incubation, not only was there a slight decrease in total cell counts but a drop in cell viability (less than 5% of active cells) and in the metabolically active population was also observed. During this period of decline (the death phase), an increase of PI-stained cells was simultaneously observed, reaching values similar to that of the CV6-stained population at 95.5 h. At this moment, 66% of bacteria failed to stain with PI, indicating that their membranes were intact, since membrane-compromised bacterial cells fluoresce in the red wavelength range.
Entry into stationary phase was probably not caused solely by carbon starvation since significant amounts of glucose (1.5 and 5.2 g · liter−1 glucose for yeast and bacterial growth, respectively) were observed after cells stopped growing. Depletion of another nutrient, cell density, or the presence of toxic products may probably induce this phase (64). Growth of lactic acid bacteria is inhibited not only by the production of metabolites such as lactic acid but also by limited concentrations of indispensable nutrients present in the MRS medium (61). It has been reported that a high level of lactic acid was one of the most likely explanations for the shortened stationary-phase persistence in Streptococcus pyogenes batch culture (65).
Using this staining protocol, ghost cells were not detected, since the presence of nucleic acids in the cells is a prerequisite for staining.
Turbidity versus FC (as total counts): kinetic assessment.
Some studies have reported that the turbidimetric method is less sensitive than plate counting (46), and its sensitivity appears to be correlated with the microbiological culture density, so it is restricted to conditions under which high cell densities are reached (10).
No statistical significant differences (P > 0.05) were found between the results obtained by turbidity and those by FC analysis for total counts (Fig. 2 and 3). The growth rate measured for each technique was taken into account. To evaluate and compare growth rates, biomass total data (g · liter−1) calculated by OD and by FC (DRAQ5 staining) were adjusted to the Riccatti model (3), given by equation 1:
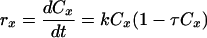 |
(1) |
where k (h−1) and τ are characteristic parameters of the system, and τ is the inverse of the cell concentration (Cx) in stationary phase expressed in g · liter−1. This empirical logistic model (equation 1) is frequently used to simulate microbial growth when the population of cells inhibits their own growth via depletion of a limiting nutrient, by production accumulation, or for undefined reasons (25). Parameters obtained for S. cerevisiae and L. hilgardii growth are shown in Table 1. The specific growth rate, μ (h−1), and the yield coefficient, Yxs (g cell·g glucose−1), were calculated as well.
TABLE 1.
Kinetic parameters for S. cerevisiae and L. hilgardii growth by dry weight and FC measurements
| Organism | Cell concn (Cx) | Parameter
|
|||
|---|---|---|---|---|---|
| μ (h−1) | τ (g · liter−1) | k (h−1) | Yxs (g cell·g glucose−1) | ||
| S. cerevisiae | Biomass total (dry wt) | 0.03 | 0.10 | 0.20 | 0.54 |
| Total cells (by FC) | 0.05 | 0.06 | 0.22 | 0.85 | |
| L. hilgardii | Biomass total (dry wt) | 0.12 | 1.20 | 0.35 | 0.05 |
| Total cells (by FC) | 0.15 | 0.53 | 0.38 | 0.12 | |
In general, specific growth rates were almost similar for dry weight and FC measurements, although the growth rates determined by FC were slightly higher. Specific growth rates for yeast growth were 0.03 and 0.05 h−1 using dry weight and FC measurements, respectively. In the case of malolactic bacteria, the values of specific growth rate found were 0.12 h−1 by dry weight and 0.15 h−1 calculated by DRAQ5 staining. Obviously, higher growth rates do not suppose that cells divide more rapidly but in this case can be explained based on the different characteristics of each method. Laplace-Builhé et al. (27) pointed out that significant differences could be obtained in the count of viable bacteria in the production of lactic starter cultures according to the enumeration method. As described above, the maximum cell concentrations were different depending on the technique used to determine total biomass, reaching higher values from FC analysis (16.8 g · liter−1 for yeast and 1.9 g · liter−1 for bacteria). The same considerations could be taken into account for the evaluation of yield coefficients. Glucose-to-cell yield coefficients (Yxs) by the FC technique took higher values than those calculated by dry weight: 0.54 compared to 0.85 g cell·g glucose−1 for yeast and 0.05 relative to 0.12 g cell·g glucose−1 for bacterial growth. The results obtained were expected since the total cell concentration calculated by FC is greater than biomass values expressed as dry weight. The estimated values of Yxs agreed with those generally found for aerobic growth of S. cerevisiae cultures (3, 23, 60) and some species of Lactobacillus in batch cultivation (29, 43, 44).
Evolution of viable and VBNC cells.
Contrary to absorbance measurements, which do not allow distinguishing between different subpopulations, FC in combination with CV6 and PI staining was useful to evaluate active and dead cells in only one staining step. Multiparametric dual-staining protocols have been applied for bacterium and yeast analysis (see references 28, 30, and 46, among other references).
Differences in cell counts obtained when applying different techniques were found, as previously referred (2, 7, 8). Note that the flow cytometric determination resulted in higher cell numbers than those determined by CFU. It should be highlighted that the principle and cell numbers considered by each technique are different. After acid challenge of Streptococcus macedonicus (41), a heterogeneity within the bacterial population was observed, which was otherwise undetermined by the averaging effect of bulk measurements or the binary logic imposed by traditional culture-based techniques. Different physiological states (showing discordances between CFU and FC counts) were detected under the stress conditions found in cider fermentation processes over time (both as pure and mixed cultures) (20). As shown in Fig. 2 and 3, the comparison between CFU and FC results showed that viable cell counts were lower than the metabolically active population throughout both growth curves. For the yeast growth curve, good correlations were obtained between viable cell counting and FC measurements for exponential and stationary phase (R2, 0.98 and 0.89, respectively). Kacmar et al. (23) found a population of nonviable cells that still had green fluorescent protein (Gfp) fluorescence during stationary phase in S. cerevisiae growing in batch cultures. In that study, the viability of several recombinant strains, determined by Gfp and PI staining, decreased at a different rate according to specific growth rates during stationary phase. As reported previously (64), stationary-phase S. cerevisiae cells are able to survive for prolonged periods and maintain nearly 100% viability for up 3 months without additional nutrients when grown in rich medium.
Similarly, the bacterial counts obtained by CV6 staining correlated with plate counts for exponential (R2, 0.90) and stationary phase (R2, 0.96). The correlation coefficient, however, cannot be interpreted directly in terms of accuracy (31). Differences between viable, active, and total bacteria were found not only in stationary phase but also in exponential growth phase (Fig. 3), as it was reported by Gallant and Palmer (18). They found that a small fraction (0.5% of the total number of cells) of an exponentially growing E. coli culture failed to produce colonies on nutrient agar plates. Ericsson et al. (17) applied viability assays of stationary phase E. coli cultures at the time of growth arrest and showed differences between CFU counts and total counts, since 100% of the original population remained intact for extended periods of time despite the loss of viability. Other studies showed a transient decrease in the number of colony-forming cells during extended stationary phase of Micrococcus luteus (24), Rhodococcus rhodochrous, and Mycobacterium tuberculosis batch cultures (55).
The evolution of VBNC cells for yeasts and bacterial growth is shown in Fig. 5. This subpopulation was calculated as CV6-stained cells minus cells capable of growing on agar plates. Desnues et al. (15) pointed out the interesting question about how asymmetry in population damage is generated. The presence of high numbers of yeast and bacterial cells that failed to grow on plates during exponential phase might be influenced in this case by the use of microbial cells after preservation forms as inoculums (a commercial active dried form and a freezing vial). It is known that preservation treatments affect cell recovery, and growth on solid medium can fail (42). In addition, it is was recently described that even in a presumed totally healthy sample as assessed by classical microbiology, there are already some cells in an “injured” or even a permeabilized state (41).
FIG. 5.

Experimental data (symbols) and predicted values (solid line) for yeast (A) and bacterial (B) growth and substrate evolution (○) are shown in the main and secondary y axes, respectively. VBNC (▪), viable (⧫), and dead (▴) cell subpopulations are expressed in g · liter−1.
The stationary phase is a condition that commonly favors cells to become VBNC (37), so that the kinetics of VBNC formation is affected by the nutritional state of the population (32). The transition from exponential growth to the stationary phase results in numerous changes in cell morphology and physical and biochemical properties (24). VBNC formation has been previously reported also for Streptococcus mutans and Streptococcus pyogenes batch pure cultures (49, 58, 65).
According to Divol and Lonvaud-Funel (16), the lower fluorescence intensity can be considered a signal of a lower vitality of the cells, resulting in the general decrease of enzymatic activity. In our experiments, taking as reference the fluorescence intensity obtained for fresh overnight cultures, CV6 fluorescence intensity was maintained during overall bacterial growth (except during death phase), so it can be considered that bacterial VBNC cells were able to preserve the whole enzymatic activity. In contrast, a slight decrease in CV6 fluorescence intensity was observed for yeasts during stationary phase.
There have been many reports which support the proposition that nonculturable cells are alive (6, 38, 39) as well as metabolically active (4, 19, 53), although VBNC cells can also demonstrate very low levels of metabolic activity (40).
Nowadays, the problem of the presence and resuscitation of VBNC cells remains unresolved. The real explanation to this phenomenon as well as the transitional stages between culturable and nonculturable states is poorly understood (13). Some authors argue that VBNC cells become progressively debilitated until cell death, so it is uncertain whether the VBNC state represents a programmed genetic response of a cell to overcoming adverse conditions or an end-of-life process (32, 57). However, what it is known is that culture conditions need to be perfect for any cell to divide and that the environmental conditions on the surface on a solid agar plate are very different to those experienced by a cell when submerged in a liquid medium in a bioreactor (22). Even, the plating medium itself may be a factor in the nonculturability associated with the VBNC state since elevated nutrient might be toxic in some manner to cells in this state, as well as high surface tension and oxygen concentration in surface growth (26, 40). At least for some cells, one aspect of the VBNC state may involve the natural presence of peroxide in solid medium, coupled with the inability of cells to detoxify this lethal metabolite (40). Therefore, it may be possible that while cells do not divide on agar plates, they may be capable of division in the bioreactor (56). In E. coli glucose-limited, fed-batch cultures, a recombinant promegaprotein was produced at high rate, although a drastic drop in the cell number of CFU·ml−1 was observed, while viability (measured as the absence of PI staining) and dry cell weight continued to increase (56).
Kinetic equations based on different physiological states and glucose consumption: a segregated model.
The total biomass was considered as formed by viable (CV), viable but nonculturable (CN), and dead cells (Cd) expressed as the cell concentration (g · liter−1). In order to develop a kinetic model, some considerations must be taken into account. In this study, the term “viable” refers to the cell subpopulation showing the ability to grow on plates, and both viable and nonculturable fractions lose their integrity, which results in cell death. In view of the results obtained and assuming that VBNC cells are not able to resuscitate, at least in this context and under the assayed conditions, three possible schemes were proposed (Fig. 4), and the reaction rates were defined as the following:
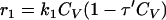 |
(2) |
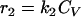 |
(3) |
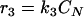 |
(4) |
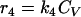 |
(5) |
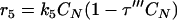 |
(6) |
Cell growth rates (r1 and r5) are described by the Riccatti equation (equation 1), where τ′ and τ‴ are the inverses of viable and VBNC cell concentrations, respectively, in stationary phase. Cell death rates (r3 and r4) respond to a first-order kinetic equation. r2 is the rate of formation of VBNC cells as a consequence of viable cell dynamics during growth and the entrance into stationary phase. First, r2 was considered as proportional to CV (equation 3), but experimental data did not fit in a suitable way. The next alternative considered r2 as a second-order reaction depending on CN. In two models proposed, r5 was equal to 0, that is, the VBNC subpopulation curve observed was only due to the entrance of viable cells into nonculturable state. Thus, there should exist some unknown factor or cell mechanism arresting the transformation into the nonculturable state at stationary phase. Finally, the last alternative proposed considered the growth of a VBNC fraction in liquid medium (equation 6) and, in addition, a first-order transformation into VBNC cells (equation 3). The growth kinetic model formed by equations 2, 3, 4, 5, and 6 was fitted to experimental data from cell counting. Figure 5 shows experimental data and predicted values for yeast and bacterial growth curves.
FIG. 4.

Schemes proposed for yeast and bacterial growth in semisynthetic medium.
Experimental data for glucose consumption were adjusted to the maintenance energy model (48). Maintenance energy is the energy required for survival or for preservation of cell viability, which is not directly related to or coupled with the synthesis of a new cell (3). Glucose is used for both cell growth and maintenance; thus, the glucose uptake rate can be expressed as:
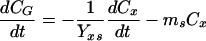 |
(7) |
Data obtained by FC analysis in combination with plate counting have been proven useful to develop a kinetic model considering different physiological states in the microbial population. The segregated model proposed describes the heterogeneity in the physiological states observed, including the VBNC fraction and dead cells, explaining more properly the cell dynamics in the bioreactor and opening the way to study the interactions between different states. Kinetic constants can be maintained under more variable conditions, and the fitting of glucose uptake data by metabolically active cells gave better correlations than those based only on viable cells.
The FC technique provides valuable information especially relating to the different metabolic activities or physiological states of individual cells when designing a bioprocess or investigating the effect of different strategies to increase the overall process efficiency.
TABLE 2.
Kinetics parameters for S. cerevisiae and L. hilgardii growth and glucose consumption
| Organism | Cell concn
|
Specific growth rate (h−1) of cells
|
Yxs (g cell·g glucose−1) | |||||
|---|---|---|---|---|---|---|---|---|
| τ′ (g · liter−1) | τ‴ (g · liter−1) | k1 | k2 | k3 | k4 | k5 | ||
| S. cerevisiae | 0.23 | 0.11 | 0.45 | 6.5 × 10−3 | 1.4 × 10−4 | 3.3 × 10−5 | 0.23 | 0.76 |
| L. hilgardii | 3.60 | 0.65 | 0.31 | 1.2 × 10−2 | 2.3 × 10−3 | 1.9 × 10−4 | 0.45 | 0.11 |
Acknowledgments
We thank Ana Salas (Flow Cytometry Area, Scientific-Technical Services, University of Oviedo) and acknowledge the support of the Asturian cider-making industry Sidra Escanciador, S.A., (Villaviciosa, Principado de Asturias, Spain).
Footnotes
Published ahead of print on 4 May 2007.
REFERENCES
- 1.Alcon, A., V. E. Santos, J. A. Casas, and F. García-Ochoa. 2004. Use of flow cytometry for growth structured kinetic model development. Application to Candida bombicola growth. Enzyme Microb. Technol. 34:399-406. [Google Scholar]
- 2.Amor, K. B., P. Breeuwer, P. Verbaarschot, F. M. Rombouts, A. D. L. Akkermans, W. M. De Vos, and T. Abee. 2002. Multiparametric flow cytometry and cell sorting for the assessment of viable, injured, and dead bifidobacterium cells during bile salt stress. Appl. Environ. Microbiol. 68:5209-5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey, J. E., and D. F. Ollis. 1986. Biochemical engineering fundamentals, 2nd ed. McGraw-Hill, Inc., New York, NY.
- 4.Barer, M. R., L. T. Gribbon, C. R. Harwood, and C. E. Nwoguh. 1993. The viable but non-culturable hypothesis and medical bacteriology. Rev. Med. Microbiol. 4:183-191. [Google Scholar]
- 5.Bley, T., S. Müller, and A. Lösche. 1995. Flow cytometry monitoring of bacterial cells states under growth limiting conditions. Comput. Appl. Biotechnol. 213-216.
- 6.Bogosian, G., and E. V. Bourneuf. 2001. A matter of bacterial life and death. EMBO Rep. 2:770-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunthof, C. J., K. Bloemen, P. Breeuwer, F. M. Rombouts, and T. Abee. 2001. Flow cytometric assessment of viability of lactic acid bacteria. Appl. Environ. Microbiol. 67:2326-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bunthof, C. J., and T. Abee. 2002. Development of a flow cytometric method to analyze subpopulations of bacteria in probiotic products and dairy starters. Appl. Environ. Microbiol. 68:2934-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cánovas, M., V. García, V. Bernal, T. Torroglosa, and J. L. Iborra. 2007. Analysis of Escherichia coli cell state by flow cytometry during whole cell catalyzed biotransformation for l-carnitine production. Process Biochem. 42:25-33. [Google Scholar]
- 10.Dalgaard, P., and K. Koutsoumanis. 2001. Comparison of maximum specific growth rates and lag times estimated from absorbance and viable count data by different mathematical models. J. Microbiol. Methods 43:183-196. [DOI] [PubMed] [Google Scholar]
- 11.Davey, H. M., and D. B. Kell. 1996. Flow cytometry and cell sorting of heterogeneous microbial populations—the importance of single-cell analyses. Microbiol. Ref. 60:641-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davey, H. M., D. H. Weichart, D. B. Douglas, and A. S. Kapreylants. 1999. Estimation of microbial viability using flow cytometry, p. 11311-11320. In J. P. Robinson, Z. Darzynkiewicz, P. N. Dean, et al. (ed.), Current protocols in cytometry. Wiley & Sons, New York, NY.
- 13.Decker, E.-M. 2001. The ability of direct fluorescence-based, two colour assays to detect different physiological sates of oral streptococci. Lett. Appl. Microbiol. 33:188-192. [DOI] [PubMed] [Google Scholar]
- 14.Deere, D., J. Porter, C. Edwards, and R. Pickup. 1995. Evaluation of suitability of bis-(1,3-dibutylbarbituric acid) trimethine oxonol (diBAC 4(3)−) for the flow cytometric assessment of bacterial viability. FEMS Microbiol. Lett. 130:165-169. [DOI] [PubMed] [Google Scholar]
- 15.Desnues, B., C. Cuny, G. Gregori, S. Dukan, H. Aguilaniu, and T. Nyström. 2003. Differential oxidative damage and expression of stress defense regulons in culturable and non-culturable Escherichia coli cells. EMBO Rep. 4:400-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Divol, B., and A. Lonvaud-Funel. 2005. Evidence for viable but non-culturable yeasts in botrytis-affected wine. J. Appl. Microbiol. 99:85-93. [DOI] [PubMed] [Google Scholar]
- 17.Ericsson, M., D. Hanstorp, D. Hagberg, J. Enger, and J. Nyström. 2000. Sorting out bacterial viability with optical tweezers. J. Bacteriol. 182:5551-5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallant, J., and L. Palmer. 1979. Error propagation in viable cells. Mech. Ageing Dev. 10:27-38. [DOI] [PubMed] [Google Scholar]
- 19.Gunasekera, T. S., A. Sorensen, P. V. Attfield, S. J. Sorensen, and D. Veal. 2002. Inducible gene expression by nonculturable bacteria in milk after pasteurization. Appl. Environ. Microbiol. 68:1988-1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrero, M., C. Quirós, L. A. García, and M. Díaz. 2006. Flow cytometry to follow the physiological states of microorganisms in cider fermentation processes. Appl. Environ. Microbiol. 72:6725-6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hewitt, C. J., and G. Nebe-von-Caron. 2001. An industrial application of multiparameter flow cytometry: assessment of cell physiological state and its application to the study of microbial fermentations. Cytometry 44:179-187. [DOI] [PubMed] [Google Scholar]
- 22.Hewitt, C. J., and G. Nebe-von-Caron. 2004. The applications of flow cytometry to monitor cell physiological state. Adv. Biochem. Eng. Biotechnol. 89:197-223. [DOI] [PubMed] [Google Scholar]
- 23.Kacmar, J., A. Zamamiri, R. Carlson, N. R. Abu-Absi, and F. Srienc. 2004. Single-cell variability in growing S. cerevisiae cell populations measured with automated flow cytometry. J. Biotechnol. 109:239-254. [DOI] [PubMed] [Google Scholar]
- 24.Kaprelyants, A. S., and D. B. Kell. 1993. Dormancy in stationary-phase cultures of Micrococcus luteus: flow cytometry analysis of starvation and resuscitation. Appl. Environ. Microbiol. 59:3187-3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kedia, G., R. Wang, H. Patel, and S. S. Pandiella. 2007. Use of mixed cultures for the fermentation of cereal-based substrates with potential probiotic properties. Process Biochem. 42:65-70. [Google Scholar]
- 26.Kell, D. B., A. S. Kaprelyants, D. H. Weichart, C. R. Harwood, and M. R. Barer. 1998. Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie Leeuwenhoek 73:169-187. [DOI] [PubMed] [Google Scholar]
- 27.Laplace-Builhé, C., K. Hahne, W. Hunger, Y. Tirilly, and J. L. Drocourt. 1993. Application of flow cytometry to rapid microbial analysis in food and drink industries. Biol. Cell 78:123-128. [DOI] [PubMed] [Google Scholar]
- 28.Lebaron, P., N. Parthuisot, and P. Catala. 1998. Comparison of blue nucleic acid dyes for flow cytometry enumeration of bacteria in aquatic systems. Appl. Environ. Microbiol. 64:1725-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lejeune, R., R. Callewaert, K. Crabbé, and L. De Vuyst. 1998. Modelling the growth and bacteriocin production by Lactobacillus amylovorous DCE 471 in batch cultivation. J. Appl. Microbiol. 84:159-168. [Google Scholar]
- 30.Lopes da Silva, T., A. Reis, C. A. Kent, M. Kosseva, J. C. Roseiro, and C. J. Hewitt. 2005. Stress-induced physiological responses to starvation period as well as glucose and lactose pulses in Bacillus licheniformis CCMI 1034 continuous aerobic fermentation processes as measured by multi-parameter flow cytometry. Biochem. Eng. J. 24:31-41. [Google Scholar]
- 31.Massart, D. L., B. G. M. Vandeginste, S. M. Deming, Y. Michotte, and L. Kaufman. 2003. Evaluation of precision and accuracy. Comparison of two procedures, p. 33-57. In Chemometrics: a textbook. Elsevier Science, B. V., Amsterdam, The Netherlands.
- 32.McDouglad, D., S. A. Rice, D. Weichart, and S. Kjelleberg. 1998. Non culturability: adaptation or debilitation? FEMS Microbiol. Ecol. 25:1-9. [Google Scholar]
- 33.Miller, G. L. 1959. Use of dinitrosalicylic acid reagent for determinination of reducing sugar. Anal. Chem. 31:426-428. [Google Scholar]
- 34.Natarajan, A., and F. Srienc. 1999. Dynamics of glucose uptake by single Escherichia coli cells. Metab. Eng. 1:320-333. [DOI] [PubMed] [Google Scholar]
- 35.Nebe-von Caron, G., and R. A. Badley. 1995. Viability assessment of bacteria in mixed populations using flow cytometry. J. Microsc. 179:55-66. [Google Scholar]
- 36.Nebe-von-Caron, G., P. J. Stephens, C. J. Hewitt, J. R. Powell, and R. A. Badley. 2000. Analysis of bacterial function by multi-colour fluorescence flow cytometry and single cell sorting. J. Microbiol. Methods 42:97-114. [DOI] [PubMed] [Google Scholar]
- 37.Nyström, T. 2005. Bacterial senescence, programmed death, and premeditated sterility. ASM News 71:363-369. [Google Scholar]
- 38.Oliver, J. D. 1993. Formation of viable but non-culturable cells, p. 239-271. In S. Kjelleberg (ed.), Starvation in bacteria. Plenum Press, New York, NY.
- 39.Oliver, J. D. 2000. The viable but nonculturable state and cellular resuscitation, p. 723-730. In C. R. Bell, M. Brylinsky, and P. Johnson-Green (ed.), Microbial biosystems: new frontiers. Atlantic Canada Society for Microbial Ecology, Halifax, Canada.
- 40.Oliver, J. D. 2005. The viable but non-culturable state in bacteria. J. Microbiol. 43:93-100. [PubMed] [Google Scholar]
- 41.Papadimitriou, K., H. Pratsinis, G. Nebe-von Caron, D. Kletsas, and E. Tsakalidou. 2006. Rapid assessment of the physiological status of Streptococcus macedonicus by flow cytometry and fluorescence probes. Int. J. Food Microbiol. 111:197-205. [DOI] [PubMed] [Google Scholar]
- 42.Parthuisot, N., P. Catala, P. Lebaron, D. Clermont, and C. Bizet. 2003. A sensitive and rapid method to determine the viability of freeze-dried bacterial cells. Lett. Appl. Microbiol. 36:412-417. [DOI] [PubMed] [Google Scholar]
- 43.Passos, F. V., H. P. Fleming, D. Ollis, R. M. Felder, and R. F. McFeeters. 1994. Kinetics and modeling of lactic acid production by Lactobacillus plantarum. Appl. Environ. Microbiol. 60:2627-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Passos, F. V., H. P. Fleming, H. M. Hassan, and R. F. McFeeters. 2003. Effect of malic acid on the growth kinetics of Lactobacillus plantarum. Appl. Microbiol. Biotechnol. 63:207-211. [DOI] [PubMed] [Google Scholar]
- 45.Peña, C., M. A. Trujillo-Roldán, and E. Galindo. 2000. Influence of dissolved oxygen tension and agitation speed on alginate production and its molecular weight in cultures of Azotobacter vinelandii. Enzyme Microb. Technol. 27:390-398. [DOI] [PubMed] [Google Scholar]
- 46.Pianetti, A., T. Falcioni, F. Bruscolini, L. Sabatini, E. Sist., and E. Papa. 2005. Determination of the viability of Aeromonas hydrophila in different types of water by flow cytometry and comparison with classical methods. Appl. Environ. Microbiol. 71:7948-7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pinder, A. C., P. W. Purdy, S. A. G. Poulder, and D. C. Clark. 1990. Validation of flow cytometry for rapid enumeration of bacterial concentrations in pure cultures. J. Appl. Bacteriol. 69:92-100. [DOI] [PubMed] [Google Scholar]
- 48.Pirt, S. J. 1965. The maintenance energy of bacteria in growing cultures. Proc. R. Soc. Lond. B 163:224-231. [DOI] [PubMed] [Google Scholar]
- 49.Renye, J. A., Jr., P. J. Piggot, L. Daneo-Moore, and B. A. Buttaro. 2004. Persistence of Streptococcus mutans in stationary-phase batch cultures and biofilms. Appl. Environ. Microbiol. 70:6181-6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rieseberg, M., C. Kasper, K. F. Reardon, and T. Scheper. 2001. Flow cytometry in biotechnology. Appl. Microbiol. Biotechnol. 56:350-360. [DOI] [PubMed] [Google Scholar]
- 51.Robertson, B. R., D. K. Button, and A. L. Koch. 1998. Determination of the biomasses of small bacteria at low concentrations in a mixture of species with forward light scatter measurements by flow cytometry. Appl. Environ. Microbiol. 64:3900-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheper, T., H. Hoffman, and K. Scügerl. 1987. Flow cytometric studies during culture of Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 9:399-405. [Google Scholar]
- 53.Shapiro, H. M. 2003. Practical flow cytometry, 4th ed. John Wiley & Sons, Hoboken, NJ.
- 54.Shleeva, M. O., K. Bragamyan, M. V. Telkov, G. V. Mukamolova, M. Young, D. B. Kell, and A. S. Kaprelyants. 2002. Formation and resuscitation of non-culturable cells of Rhodococcus rhodochrous and Mycobacterium tuberculosis in prolonged stationary phase. Microbiology 148:1581-1591. [DOI] [PubMed] [Google Scholar]
- 55.Stockall, A. M., and C. Edwards. 1985. Changes in respiratory activity during encystment of Azotobacter vinelandii. J. Gen. Microbiol. 131:1403-1410. [Google Scholar]
- 56.Sundström, H., F. Wallberg, E. Ledung, B. Norrman, C. K. Hewitt, and S.-O. Enfors. 2004. Segregation to non-dividing cells in recombinant Escherichia coli fed-batch fermentation processes. Biotechnol. Lett. 26:1533-1539. [DOI] [PubMed] [Google Scholar]
- 57.Trainor, V. C., R. K. Udy, P. J. Bremer, and G. M. Cook. 1999. Survival of Streptococcus pyrogenes under stress and starvation. FEMS Microbiol. Lett. 176:421-428. [DOI] [PubMed] [Google Scholar]
- 58.Van Hoek, P., J. P. van Dijken, and J. T. Pronk. 1998. Effect of specific growth rate on fermentative capacity of baker's yeast. Appl. Environ. Microbiol. 64:4226-4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Veal, D. A., D. Deere, B. Ferrari, J. Piper, and P. V. Attfield. 2000. Fluorescence staining and flow cytometry for monitoring microbial cells. J. Immunol. Methods 243:191-210. [DOI] [PubMed] [Google Scholar]
- 60.Verduyn C., A. H. Stouthamer, W. A. Scheffers, and van J. P. Dijken. 1991. A theoretical evaluation of growth yields of yeasts. Antonie Leeuwenhoek 59:49-63. [DOI] [PubMed] [Google Scholar]
- 61.Verluyten, J., F. Leroy, and L. Vuyst. 2004. Influence of complex nutrient source on growth of and Curvacin A production by sausage isolate Lactobacillus curvatus LTH 1174. Appl. Environ. Microbiol. 70:5081-5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walker, G. M. 1999. Synchronization of yeast cell populations. Methods Cell Sci. 21:87-93. [DOI] [PubMed] [Google Scholar]
- 63.Wallberg, F. 2004. Flow cytometry for bioprocess control. Ph.D. dissertation. Royal Institute of Technology, Stockholm, Sweden.
- 64.Werner-Washburne, M., E. Braun, G. C. Johnston, and R. A. Singer. 1993. Stationary phase in the yeast Saccharomyces cerevisiae. Microbiol. Rev. 57:383-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wood, D. N., M. A. Chaussee, M. S. Chaussee, and B. A. Buttaro. 2005. Persistence of Streptococcus pyogenes in stationary-phase cultures. J. Bacteriol. 187:3319-3328. [DOI] [PMC free article] [PubMed] [Google Scholar]