

Abstract
Retinitis pigmentosa (RP) is a group of progressive retinal dystrophies that include the most common hereditary degenerative diseases affecting the retina. Although most disease phenotypes appear to result from defects at single genetic loci (monogenic), at least one instance of RP appears to require a coinheritance of defects in the unlinked peripherin/rds and rom-1 alleles (digenic), which encode the polypeptide subunits of an oligomeric transmembrane protein complex present at photoreceptor outer segment disc rims. Sedimentation velocity analysis was performed upon the affected gene products expressed heterologously in COS-1 cells to examine the assembly of the subunit polypeptides. The results indicate that the missense peripherin/rds mutant, L185P, which segregates with instances of digenically inherited RP, is conditionally defective with respect to its subunit assembly. Unlike wild-type peripherin/rds, the L185P mutant does not form native-like homotetramers on its own; however, the L185P mutant can assemble with wild-type rom-1 to form a structurally normal heterotetrameric complex. These findings provide a novel molecular-based rationale for the unusual digenic disease inheritance pattern and offer insight into regions of peripherin/rds and rom-1, which contribute to subunit–subunit interactions.
Retinitis pigmentosa (RP) is a heterogeneous group of inherited retinal diseases that affects an estimated 1.5 million individuals worldwide. Typical symptoms include night blindness, a progressive reduction in peripheral visual field and subsequent loss of central vision, the presence of subretinal pigmentary patches, a pale optic disc, attenuation of retinal vessels, and abnormal electroretinograms in advanced stages of the disease (1, 2, 3, 4, 5). RP is genetically heterogeneous; pathogenicity has been linked both to multiple loci (allelic heterogeneity) and to different mutations at particular loci (nonallelic heterogeneity) (2, 3, 4, 5). Currently, seven genes highly expressed in photoreceptor cells have been linked to various forms of autosomal dominant and autosomal recessive RP (5). They encode key proteins in the phototransduction cascade: rhodopsin, the α and β-subunits of cGMP-phosphodiesterase, and the α-subunit of the cGMP-gated cation channel, as well as proteins required for normal photoreceptor outer segment morphogenesis and structure, peripherin/rds and rom-1. Defects in the genes for rhodopsin and peripherin/rds have been most extensively documented: over 70 mutations in rhodopsin and over 30 mutations in peripherin/rds collectively account for ≈30% of individuals with autosomal dominant RP (5). The molecular mechanism(s) by which mutations in these genes lead to photoreceptor degeneration remains to be elucidated. In the case of rhodopsin, however, certain mutations have been shown to result in misfolded or functionally altered proteins; in some instances, targeting to the plasma membrane of cultured cells and the outer segments of photoreceptor cells is disrupted (6, 7, 8, 9, 10). The effects of disease-linked mutations on peripherin/rds structure and cellular distribution have yet to be reported.
Peripherin/rds is an integral membrane glycoprotein that, together with its nonglycosylated homologue rom-1, forms a multisubunit complex at the rim region of rod and cone outer segment discs (11, 12, 13, 14, 15, 16). Peripherin/rds appears to be essential for the formation and maintenance of normal photoreceptor outer segments since a disruptive insertion of foreign DNA into the RDS gene is responsible both for the inability of homozygous retinal degenerative slow (rds) mice to develop photoreceptor outer segments and for the disorganized, unstable outer segments found in heterozygous rds mice (17, 18, 19, 20). In humans, mutations in peripherin/rds have been linked to a variety of progressive retinal degenerations, including autosomal dominant RP (21, 22, 23, 24, 25, 26, 27, 28), retinitis punctata albescens (29), macular dystrophies (26, 28, 30), and fundus flavimaculatus (28).
Peripherin/rds and rom-1 have also been implicated in at least one instance of a recently documented polygenic retinal disease (31). In this pedigree, only those family members who coinherit the peripherin/rds missense mutation, L185P, and an apparently null mutation in the gene coding for rom-1 (double heterozygotes) exhibit a disease phenotype. Individuals who carry either one or the other of these mutations (single heterozygotes) appear to be essentially normal. The resultant inheritance pattern is distinct from both monogenic dominant and recessive modes and is known as digenic (5, 31). Since disulfide-linked homodimers of peripherin/rds are known to interact with disulfide-linked homodimers of rom-1 to form a multimeric transmembrane complex (15, 16, 32), we have investigated whether subunit assembly plays a role in this particular type of autosomal dominant RP.
MATERIALS AND METHODS
Expression Plasmids.
Individual plasmids are based on the pcDNAI/AMP (Invitrogen) mammalian expression vector and contain full-length coding regions for wild-type (WT) bovine peripherin/rds (pcPER) and rom-1 (pcROM) as described (33). Missense peripherin/rds mutations L185P and R172W (26, 31) were generated by site-directed mutagenesis of the pcPER plasmid with a Muta-Gene Phagmid kit (Bio-Rad) using the sense-strand synthetic oligonucleotides: 5′-CCGCTATCCGGATTTTTCC-3′ (L185P) and 5′-ACGGCTTTTGGGACTGG-3′ (R172W). The mutagenized plasmids were verified by complete (single-strand) dideoxy-sequencing with a Sequenase T7 kit (United States Biochemical). Since the rom-1 mutations implicated in digenic RP (G80ins and L114ins) are frameshifts that result in premature termination at codon 131, they are considered to be null alleles (31) and were not constructed for this study.
Heterologous Expression.
COS-1 cells (ca. 2 × 105 per 60-mm dish) were singly transfected with one of the peripherin/rds expression plasmids (12 μg of pcPER, pcR172W, or pcL185P) or cotransfected with a mixture of 6 μg of pcROM and 6 μg of one of the peripherin/rds expression plasmids (above), then solubilized with 1% Triton X-100 in phosphate-buffered saline 72 h posttransfection, as described (33). Procedures for the assay of expression and dimerization by Western blotting and for the assembly of peripherin/rds–rom-1 complexes by coprecipitation have been reported (33).
Velocity Sedimentation.
Peripherin/rds-containing complexes were immunoaffinity-purified from transfected COS-1 cell lysates with anti-peripherin monoclonal antibody per2B6 covalently linked to Sepharose beads and sedimented through 5–20% sucrose gradients as reported (33). Sedimentation profiles of assembled peripherin/rds homomeric and peripherin/rds–rom-1 heteromeric complexes were determined by chemiluminescent Western blotting (using anti-peripherin/rds monoclonal antibody per2B6 or anti-rom-1 polyclonal antibody romC2) and laser densitometry according to ref. 33. Peripherin/rds–rom-1 assembled complexes were additionally identified by a sequentially reversed protocol: immunoaffinity purification using an anti-rom-1 monoclonal antibody rom1D5 (16) followed by Western blot analysis with the anti-peripherin monoclonal antibody per2B6; identical results were obtained.
Sedimentation coefficients estimated initially (33) assumed a partial specific volume (ν̄) of 0.8 ml/g. A hydrodynamic characterization of the peripherin/rds–rom-1 complex from bovine rod outer segments subsequently indicated a value of ν̄ = 0.83 ml/g (32). S20,w values in this report use the experimentally determined ν̄ = 0.83 ml/g and are in good agreement with our previous reports (32, 33).
RESULTS
Expression and Dimerization of Peripherin/rds Mutants.
Previous studies have established that WT peripherin/rds expressed in COS-1 cells is essentially similar to that found in the disc membranes of photoreceptor outer segments (33). Fig. 1A compares the heterologous expression of WT peripherin/rds with two disease-linked mutants: (i) the L185P mutant involved in digenic RP (31) and (ii) a R172W mutation that segregates (monogenically) with an autosomal dominant form of macular dystrophy (26). All three peripherin/rds variants display similar electrophoretic mobilities and expression levels when detergent extracts from transiently transfected COS-1 cells are subjected to SDS/PAGE under reducing conditions. Each also contains N-linked carbohydrate as indicated by an increased electrophoretic mobility upon endoglycosidase treatment (not shown). When WT peripherin/rds is solubilized and electrophoresed under nonreducing conditions, it migrates at approximately twice the molecular weight of its monomer form (70 versus 35 kDa), suggesting that it is present primarily in the form of disulfide-linked dimers, akin to peripherin/rds from outer segment membranes (11, 12). Under identical conditions, the L185P mutant migrates as a more heterogeneous smear (Fig. 1B), suggesting that protein aggregation occurs, possibly mediated through secondary sulfhydryl oxidation processes. Inclusion of iodoacetamide or N-ethylmaleimide in cell membrane solubilization buffers appears to increase slightly the proportion of L185P migrating in the dimer mobility range (not shown). These results indicate that although the L185P mutant is expressed in COS-1 cells as a full-length polypeptide (at approximately WT levels), it also appears more susceptible to aggregation in a denaturing/nonreductive environment and may be somewhat misfolded or incorrectly assembled. It is unclear from these experiments whether this mutant is competent to form disulfide-linked homodimers. In contrast, the R172W mutant appears entirely normal by this criterion.
Figure 1.
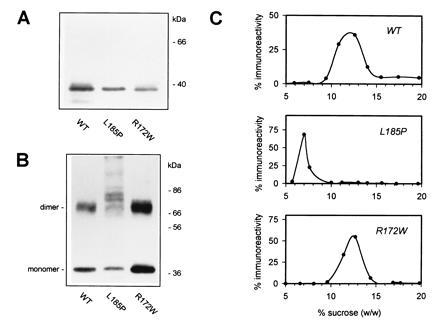
Expression and oligomerization of WT and mutant peripherin/rds. COS-1 cells transfected with the indicated expression plasmids (pcPER, pcR172W, or pcL185P) were detergent-solubilized 72 h posttransfection and probed by Western blot analysis with anti-peripherin/rds monoclonal antibody per2B6. Approximately 1.5 μg of total protein per lane was subjected to SDS/PAGE in the presence (A) or absence (B) of reducing agent (2% 2-mercaptoethanol). (C) Sedimentation velocity analysis of WT and mutant peripherin/rds complexes. Immunoaffinity-purified peripherin/rds variants from transfected COS-1 cells were sedimented through 5–20% sucrose gradients and sedimentation profiles were determined by chemiluminescent Western blotting (with anti-peripherin/rds per2B6) and laser densitometry as described. In the absence of rom-1, WT peripherin/rds is assembled as a homotetramer in COS-1 cells (32, 33).
Homomeric Assembly of Peripherin/rds Mutants.
Heterologously expressed WT peripherin/rds has previously been shown to form homotetrameric structures in the absence of rom-1; the homomeric form is suggested to be present in individuals possessing a null mutation in rom-1 (32, 33). The L185P and R172W peripherin/rds mutants were examined for their ability to form homotetramers in COS-1 cells. Sedimentation velocity analyses for heterologously expressed WT and the mutant peripherin/rds variants were performed in sucrose density gradients; each variant sediments as a distinct hydrodynamic species (Fig. 1C). WT peripherin/rds and the R172W mutant each form homotetrameric complexes; however, the reduced mobility of the digenic L185P mutant indicates that it does not form normal homotetramers. In fact, it sediments closer to what is expected for a dimeric form.† Estimated sedimentation coefficients for the homomeric peripherin/rds complexes are given in Table 1. These results demonstrate that the digenic L185P mutant is not assembled into a homotetrameric form in the absence of rom-1—in sharp contrast to WT behavior. Alteration in sedimentation velocity is not a necessary consequence of a disease-linked mutation in the peripherin/rds gene, as the R172W peripherin/rds mutant, which results in macular degeneration (26), has a sedimentation coefficient indistinguishable from WT. These observations suggest that the L185P mutation adversely affects peripherin/rds subunit assembly; in particular, this mutant does not assume a homotetrameric form in the absence of rom-1.
Table 1.
Sedimentation coefficients of peripherin/rds and peripherin/rds–rom-1 complexes
| Strain | S20,w
|
Associated phenotype | |
|---|---|---|---|
| −rom-1 | +rom-1 | ||
| WT | 6.86 ± 0.17 (n = 3) | 6.39 ± 0.46 (n = 4) | Normal |
| L185P | 4.70 ± 0.24 (n = 3) | 6.55 ± 0.26 (n = 3) | RP |
| R172W | 6.97 ± 0.24 (n = 4) | 6.24 ± 0.16 (n = 3) | Macular dystrophy |
The velocity sedimentation of immunoaffinity-purified protein complexes in 5–20% sucrose density gradients (see Figs. 1C and 2C) was used to estimate sedimentation coefficients as described previously (32, 33). Homotetramers of peripherin/rds sediment slightly faster than peripherin/rds–rom-1 heterotetramers, due to the individual polypeptide molecular weights: peripherin/rds, 39 kDa; rom-1, 37 kDa.
Heteromeric Assembly of Peripherin/rds Mutants with rom-1.
Since WT peripherin/rds and rom-1 are normally assembled as a heteromeric complex in both rod outer segment disc and COS-1 cell membranes (15, 16, 32, 33), the peripherin/rds mutants were assayed for their ability to assemble with WT rom-1. Western blot analyses performed on detergent extracts of COS-1 cells cotransfected with WT rom-1 and WT, L185P, or R172W peripherin/rds show roughly similar expression levels of rom-1 and the individual peripherin/rds variants (Fig. 2A). A coprecipitation assay was used to determine whether rom-1 was associated with the individual peripherin/rds variants. In each instance, immunoprecipitation of the expressed peripherin/rds (by the peripherin/rds-specific monoclonal antibody per2B6) resulted in coprecipitation of rom-1 (Fig. 2B Right). Moreover, peripherin/rds is quantitatively immunoprecipitated in each case—no reactivity is seen in the unbound fractions (Fig. 2B Left). These results indicate that each of the mutants is assembled with rom-1 in COS-1 cells to form a heteromeric complex.
Figure 2.
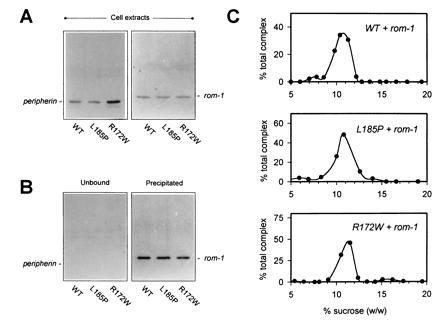
Assembly of WT and mutant peripherin/rds–rom-1 oligomeric complexes. (A) Detergent extracts from COS-1 cells cotransfected with plasmids encoding rom-1 (6 μg, pcROM) and the indicated peripherin/rds variant (6 μg) were assayed for the expression of each protein by Western blot analysis with either anti-peripherin/rds monoclonal antibody per2B6 (Left) or anti-rom-1 polyclonal antibody romC2 (Right). (B) A sequential immunoprecipitation (anti-peripherin)/Western blot (anti-rom-1) analysis was performed to assay assembly of rom-1 with each of the peripherin/rds variants. Peripherin/rds was quantitatively immunoprecipitated; in each instance, no reactivity is seen in the unbound fractions (Left). Rom-1 is observed to coprecipitate in each instance indicating heteromeric assembly with each of the mutants (Right). (C) Sedimentation velocity analysis of WT and mutant peripherin/rds–rom-1 complexes. Peripherin/rds-containing complexes were immunoaffinity-purified from transfected COS-1 cells and sedimented through 5–20% sucrose gradients. Sedimentation profiles of assembled heteromeric complexes were determined by chemiluminescent Western blotting with an anti-rom-1 antibody and laser densitometry as described (33).
The velocity sedimentation behavior for each of the coexpressed and immunopurified complexes was characterized to determine their oligomeric states (Fig. 2C). Coexpression of WT peripherin/rds with rom-1 results in the assembly of a distinct hydrodynamic species, previously shown to be tetrameric (32, 33). Like WT, the R172W mutant also forms normal heterotetramers with rom-1. Most interestingly, when the L185P digenic mutant is coexpressed with rom-1, it too is incorporated into a heterotetrameric complex, which sediments at the WT rate. These data indicate that WT rom-1 can recruit the digenic L185P peripherin/rds mutation into a normally sedimenting, presumably functional, heterotetrameric complex. Sedimentation coefficients for the peripherin/rds–rom-1 assembled complexes are summarized in Table 1.
DISCUSSION
This study indicates that the L185P mutation in the large intradiscal loop of peripherin/rds prevents the assembly of disulfide-linked peripherin/rds homodimers into homotetrameric complexes. The L185P mutant does, however, associate with disulfide-linked WT rom-1 homodimers to form heterotetrameric complexes, which are similar to those formed by WT peripherin/rds and rom-1.
These results can be incorporated into a novel model to explain the previously reported digenic RP inheritance pattern associated with mutations in peripherin/rds and rom-1 (31). This model emphasizes the functional importance of peripherin/rds-containing hetero- and homotetramers and suggests that an insufficiency of these species will lead to outer segment instability and eventual photoreceptor degeneration. As depicted in Fig. 3, peripherin/rds and rom-1 disulfide-linked homodimers associate with each other to form a normal complement of heterotetrameric complexes in individuals homozygous for WT peripherin/rds and rom-1. Heterozygous inheritance of a L185P peripherin/rds mutation in itself does not produce deleterious effects, since normal levels of rom-1 effectively recruit L185P peripherin/rds into heterotetrameric complexes as demonstrated in the current study. Individuals who are heterozygous for a rom-1 null mutation will also be relatively unaffected‡; in the absence of a full complement of rom-1, assembly of WT peripherin/rds into homotetrameric complexes would act to compensate for reduced levels of the preferred peripherin/rds–rom-1 heterotetrameric complexes. Finally, since L185P peripherin/rds appears unable to form a homotetrameric complex, a heterozygous coinheritance of the L185P peripherin/rds mutation with a rom-1 null mutation is predicted to result in a significant decrease in the abundance of peripherin/rds-containing tetramers, and an ensuing RP disease phenotype. In the simplest case, double heterozygosity for these mutations would result in haploinsufficiency of “functional” (i.e.—peripherin/rds-containing) tetrameric complexes—a condition which produces unstable outer segments in heterozygous rds mice (20) and retinal degeneration in humans heterozygous for a peripherin/rds null mutation (27, 29, 36). The presence of the improperly assembled L185P mutant also may contribute a dominant negative effect on the outer segment disc structure; though, this remains to be determined. Although the present model does not treat heterozygosity in digenic RP explicitly, a system for examining assembly of the L185P peripherin/rds mutant with WT peripherin/rds is currently being developed.
Figure 3.
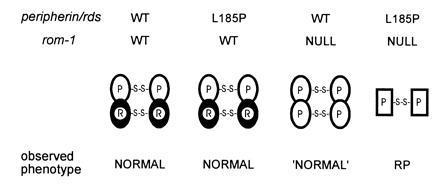
A molecular rationale for digenic RP involving the L185P peripherin/rds mutation and a rom-1 null mutation. This model stresses the functional importance of peripherin/rds-containing tetramers; instabilities (leading to RP disease phenotypes) are predicted in photoreceptors lacking sufficient levels of these particular species. Unaffected (WT) individuals and heterozygotes for a L185P mutation are expected to have normal levels of peripherin/rds–rom-1 heterotetramers, resulting in stable outer segments and an essentially normal phenotype. Heterozygotes for a null rom-1 mutation are expected to be borderline normal (“normal”) since peripherin/rds homotetramers can partially compensate for reduced levels of peripherin/rds–rom-1 heterotetramers. Double heterozygotes for the L185P peripherin/rds mutation and a null rom-1 mutation exhibit the RP phenotype. Reduced levels of the peripherin/rds–rom-1 heterotetramer coupled with the inability of L185P to form homotetramers will likely lead to disorganized and unstable outer segments.
The current scheme also predicts how these RP-linked mutations in peripherin/rds and rom-1 may be manifested in transgenic mice. Mice that coinherit the L185P peripherin/rds mutation with a rom-1 null mutation (double heterozygotes) are predicted to exhibit disorganized outer segments and undergo retinal degeneration, yet individuals with either one or the other of these mutations (single heterozygotes) are expected to have essentially normal outer segments and experience little if any photoreceptor degeneration (for reasons described above). Mice homozygous for the L185P peripherin/rds mutation should also develop photoreceptor outer segments; a full complement of WT rom-1 is expected to associate with the L185P polypeptides to produce normal levels of heterotetramers. In this case, however, retinal degeneration may ensue if the L185P peripherin/rds–rom-1 complex is less effective than the WT complex at stabilizing outer segment structure. Indeed, the borderline normal phenotype reported by Kajiwara et al. (31) for individuals heterozygous for the L185P perpherin/rds mutation suggests that the L185P peripherin/rds–rom-1 complex may not completely substitute for the WT complex. Mice homozygous for a rom-1 null allele are also predicted to develop outer segments, in contrast to homozygous rds mice, since peripherin/rds homotetramers can at least partially compensate for the absence of heterotetramers. However, the outer segments in such mice would be expected to have a disordered appearance, similar to those observed for (haploinsufficient) heterozygous rds mice (20), due to insufficient levels (haploinsufficient in the simplest instance), and reduced efficacy of peripherin/rds-containing tetramers required for the elaboration and stabilization of outer segment structure.
The expression and assembly properties of a R172W peripherin/rds mutant linked to instances of macular dystrophy have also been characterized. Since the primary defect in this type of degeneration appears to affect only a subset of the retinal photoreceptors (cones), structural perturbations are expected to be less global (relative to defects affecting the entire photoreceptor population). Indeed, as found in the current study, the R172W mutant appears normal with respect to expression, dimerization, glycosylation, and subunit assembly. These results suggest that R172W peripherin/rds–rom-1 complexes can form in vivo and that a relatively subtle change in their structure undermines their function in cone, but not rod, photoreceptors. Alternatively, if a cone-specific rom-1 homologue exists (15, 16), it may interact with the R172W mutant abnormally to result in a cone-initiated retinal degeneration.
This report provides a protein-level rationale for a digenic pattern of retinal disease inheritance which results from a L185P mutation in peripherin/rds and a null mutation in rom-1. It further demonstrates the utility of a mammalian cell expression system for evaluating the effect of disease-linked mutations on the molecular properties of these proteins and emphasizes the importance of subunit interactions for the manifestation of this particular form of RP. It is possible that other instances of RP associated with these proteins may also be related to defects in subunit assembly.
Acknowledgments
This work has been supported by grants from the National Eye Institute (EY06417), the Medical Research Council, and the RP Research Foundation.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: RP, retinitis pigmentosa; WT, wild type.
The sedimentation velocity of an integral membrane (detergent-binding) protein is a function of its molecular mass, Stokes radius, and detergent-binding properties; therefore, a mobility change cannot be rigorously interpreted in terms of subunit stoichiometry alone.
References
- 1.Heckenlively J R. Retinitis Pigmentosa. Philadelphia: Lippincott; 1988. [Google Scholar]
- 2.Berson E L. Invest Ophthalmol Vis Sci. 1993;34:1659–1676. [PubMed] [Google Scholar]
- 3.Shastry B S. Am J Med Genet. 1994;52:467–474. doi: 10.1002/ajmg.1320520413. [DOI] [PubMed] [Google Scholar]
- 4.Daiger S P, Sullivan L S, Rodriguez J A. Behav Br Sci. 1995;18:452–467. [Google Scholar]
- 5.Dryja T P, Li T. Hum Mol Genet. 1995;4:1739–1743. doi: 10.1093/hmg/4.suppl_1.1739. [DOI] [PubMed] [Google Scholar]
- 6.Doi T, Molday R S, Khorana H G. Proc Natl Acad Sci USA. 1990;87:4991–4995. doi: 10.1073/pnas.87.13.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sung C-H, Schneider B, Agarwal N, Papermaster D S, Nathans J. Proc Natl Acad Sci USA. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sung C-H, Davenport C M, Nathans J. J Biol Chem. 1993;268:26645–26649. [PubMed] [Google Scholar]
- 9.Olsson J E, Gordon J W, Pawlyk B S, Roof D, Hayes A, Molday R S, Mukai S, Cowley G S, Berson E L, Dryja T P. Neuron. 1992;9:815–830. doi: 10.1016/0896-6273(92)90236-7. [DOI] [PubMed] [Google Scholar]
- 10.Sung C-H, Makino C, Baylor D, Nathans J. J Neurosci. 1994;14:5818–5833. doi: 10.1523/JNEUROSCI.14-10-05818.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molday R S, Hicks D, Molday L L. Invest Ophthalmol Vis Sci. 1987;28:50–61. [PubMed] [Google Scholar]
- 12.Connell G J, Molday R S. Biochemistry. 1990;29:4691–4698. doi: 10.1021/bi00471a025. [DOI] [PubMed] [Google Scholar]
- 13.Arikawa K, Molday L L, Molday R S, Williams D S. J Cell Biol. 1992;116:659–667. doi: 10.1083/jcb.116.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molday R S. Prog Retinal Eye Res. 1994;13:271–299. [Google Scholar]
- 15.Bascom R A, Manara S, Collins L, Molday R S, Kalnins V I, McInnes R R. Neuron. 1992;8:1171–1184. doi: 10.1016/0896-6273(92)90137-3. [DOI] [PubMed] [Google Scholar]
- 16.Moritz O L, Molday R S. Invest Ophthalmol Vis Sci. 1996;37:352–361. [PubMed] [Google Scholar]
- 17.Travis G H, Brennan M B, Danielson P E, Kozak C, Sutcliffe J G. Nature (London) 1989;338:70–73. doi: 10.1038/338070a0. [DOI] [PubMed] [Google Scholar]
- 18.Travis G H, Groshan K R, Lloyd M, Bok D. Neuron. 1992;9:113–119. doi: 10.1016/0896-6273(92)90226-4. [DOI] [PubMed] [Google Scholar]
- 19.Sanyal S, Jansen H G. Neurosci Lett. 1981;21:23–26. doi: 10.1016/0304-3940(81)90051-3. [DOI] [PubMed] [Google Scholar]
- 20.Hawkins R K, Jansen H G, Sanyal S. Exp Eye Res. 1985;41:701–720. doi: 10.1016/0014-4835(85)90179-4. [DOI] [PubMed] [Google Scholar]
- 21.Farrar G J, Kenna P, Jordan S A, Kumar-Singh R, Humphries M M, Sharp E M, Sheils D M, Humphries P. Nature (London) 1991;354:478–481. doi: 10.1038/354478a0. [DOI] [PubMed] [Google Scholar]
- 22.Kajiwara K, Hahn L B, Mukai S, Travis G H, Berson E L, Dryja T P. Nature (London) 1991;354:480–483. doi: 10.1038/354480a0. [DOI] [PubMed] [Google Scholar]
- 23.Farrar G J, Kenna P, Jordan S A, Kumar-Singh R, Humphries M M, Sharp E M, Sheils D, Humphries P. Genomics. 1992;14:805–807. doi: 10.1016/s0888-7543(05)80193-4. [DOI] [PubMed] [Google Scholar]
- 24.Saga M, Mashima Y, Akeo K, Oguchi Y, Kudoh J, Shimizu N. Hum Genet. 1993;92:519–521. doi: 10.1007/BF00216463. [DOI] [PubMed] [Google Scholar]
- 25.Grüning G, Millan J M, Meins M, Beneyto M, Caballero M, Apfelstedt-Sylla E, Bosch R, Zrenner E, Prieto F, Gal A. Hum Mutat. 1994;3:321–323. doi: 10.1002/humu.1380030326. [DOI] [PubMed] [Google Scholar]
- 26.Wells J, Wroblewski J, Keen J, Inglehearn C, Jubb C, Eckstein A, Jay M, Arden G, Bhattacharya S, Fitzke F, Bird A. Nat Genet. 1993;3:213–218. doi: 10.1038/ng0393-213. [DOI] [PubMed] [Google Scholar]
- 27.Meins M, Grüning G, Blankenagel A, Krastel H, Reck B, Fuchs S, Schwinger E, Gal A. Human Mol Genet. 1993;2:2181–2182. doi: 10.1093/hmg/2.12.2181. [DOI] [PubMed] [Google Scholar]
- 28.Weleber R G, Carr R E, Murphey W H, Sheffield V C, Stone E M. Arch Ophthalmol. 1993;111:1531–1542. doi: 10.1001/archopht.1993.01090110097033. [DOI] [PubMed] [Google Scholar]
- 29.Kajiwara K, Sandberg M A, Berson E L, Dryja T P. Nat Genet. 1993;3:208–212. doi: 10.1038/ng0393-208. [DOI] [PubMed] [Google Scholar]
- 30.Nichols B E, Sheffield V C, Vandenburgh K, Drack A V, Kimura A E, Stone E M. Nat Genet. 1993;3:202–207. doi: 10.1038/ng0393-202. [DOI] [PubMed] [Google Scholar]
- 31.Kajiwara K, Berson E L, Dryja T P. Science. 1994;264:1604–1607. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- 32.Goldberg A F X, Molday R S. Biochemistry. 1996;35:6144–6149. doi: 10.1021/bi960259n. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg A F X, Moritz O L, Molday R S. Biochemistry. 1995;34:14213–14219. doi: 10.1021/bi00043a028. [DOI] [PubMed] [Google Scholar]
- 34.Bascom R A, Liu L, Heckenlively J R, Stone E M, McInnes R R. Hum Mol Genet. 1995;4:1895–1902. doi: 10.1093/hmg/4.10.1895. [DOI] [PubMed] [Google Scholar]
- 35.Sakuma H, Inana G, Murakami A, Yajima T, Weleber R G, Murphey W H, Gass D M, Hotta Y, Hayakawa M, Fujiki K, Gao Y Q, Danciger M, Farber D, Cideciyan A V, Jacobson S G. Genomics. 1995;27:384–386. doi: 10.1006/geno.1995.1066. [DOI] [PubMed] [Google Scholar]
- 36.Jacobson S G, Cideciyan A V, Kemp C M, Sheffield V C, Stone E M. Invest Ophthalmol Vis Sci. 1996;37:1662–1674. [PubMed] [Google Scholar]