

Abstract
Salmonella enterica has evolved a type III protein secretion system that allows these enteropathogens to translocate effector molecules directly into the host cell cytoplasm. These effectors mediate a variety of responses, including cytoskeletal rearrangements, cytokine production, and in certain cells, the induction of apoptosis. We report here the characterization of a substrate of this secretion system in S. enterica serovar typhimurium (Salmonella typhimurium) that is homologous to the SopE protein of Salmonella dublin implicated in bacterial entry into cultured epithelial cells. The sopE locus is located within a cluster of genes that encode tail and tail fiber proteins of a cryptic P2-like prophage, outside of the centisome 63 pathogenicity island that encodes the invasion-associated type III secretion system. Southern hybridization analysis revealed that sopE is present in only a subset of S. enterica serovars and that the flanking bacteriophage genes are also highly polymorphic. Encoding effector proteins that are delivered through type III secretion systems in highly mobile genetic elements may allow pathogens to adapt rapidly by facilitating the assembly of an appropriate set of effector proteins required for successful replication in a new environment.
Keywords: bacterial pathogenesis, horizontal gene transfer, pathogenicity island
Salmonella enterica can cause diseases that range from self-limiting gastroenteritis (e.g., food poisoning) to systemic enteric infections (e.g., typhoid fever). The type of disease is largely determined by the species of the infected host and/or the serovar of the infecting bacteria. For example, Salmonella enterica serovar typhimurium (Salmonella typhimurium) causes a typhoid-like systemic disease in mice, whereas in humans it generally causes self-limiting gastroenteritis. Of the more than 2,000 serovars of Salmonella enterica, some display a marked host specificity, whereas others can infect a wide range of hosts.
S. enterica has evolved a variety of mechanisms to colonize, replicate, and survive within the animal host. Some of these mechanisms depend on the function of at least two specialized type III protein secretion systems encoded at centisomes 31 and 63 of the Salmonella chromosome (1–3). This type of protein secretion system has also been identified in a number of other animal as well as plant pathogenic bacteria (reviewed in ref. 4). It is believed that the main function of these systems is to direct the translocation of effector proteins into host cells. Indeed, the type III secretion system encoded at centisome 63 of the Salmonella chromosome directs the translocation of several bacterial proteins into the host cell (5–7). These proteins activate host cell signaling pathways leading to a variety of responses, such as the reorganization of the actin cytoskeleton, resulting in bacterial internalization, the stimulation of nuclear responses, leading to cytokine production, and the triggering of programmed cell death in macrophages (reviewed in ref. 4).
Although the components of the type III secretion apparatus itself are well conserved among different Gram-negative bacteria, the substrates of this system so far identified appear much more diverse (4). The heterogeneity of the secreted effector proteins may be a consequence of the adaptation of each pathogen to its special niche.
Typically, the genes encoding structural components and substrates of type III secretion systems are organized in clusters within virulence-associated plasmids or pathogenicity islands (reviewed in refs. 4 and 8). This observation, in conjunction with the finding that the nucleotide composition of these genes is often distinct from that of the chromosome of their respective bacterial hosts, has led to the proposal that these systems were acquired in block by horizontal transmission (8–11). Consistent with this hypothesis, sequences resembling mobile DNA elements have often been found in the vicinity of these pathogenicity islands. For example, sequences similar to the insertion sequence IS3 are present in the vicinity of the centisome 63 pathogenicity island of Salmonella choleraesuis, Salmonella senftenberg, and Salmonella litchfield (12, 13).
Here, we describe the identification of a target of the centisome 63 type III secretion system of S. typhimurium that is encoded within the genome of a cryptic bacteriophage, outside the pathogenicity island encoding the structural components of its cognate type III secretion system. We discuss the implication of these findings for the ability of bacteria bearing functional type III secretion systems to rapidly adapt to novel environments.
MATERIALS AND METHODS
Bacterial Strains and Culture Conditions.
The wild-type S. typhimurium strain SL1344 (14) and the isogenic derivatives carrying nonpolar mutations in invG (SB161) (15), invC (SB566) (16) , sipA (SB225) (17), sipB (SB169), sipC (SB220) (18), sipD (SB221) (17), sptP (SB237) (19), spaO (SB302) (20), and invJ (SB303) (21) have been described elsewhere. Other Salmonella enterica serovars were from our laboratory collection and have been obtained from different sources. All Salmonella strains were grown in L broth under conditions that stimulate the expression of the components and targets of the invasion-associated protein secretion system encoded at centisome 63 of the Salmonella chromosome as described elsewhere (22).
Identification of S. typhimurium Secreted Proteins.
To identify some of the uncharacterized secreted proteins of S. typhimurium, we made use of strain SB221, which carries a nonpolar null mutation in sipD. This mutation results in increased secretion of proteins through the centisome 63 type III system (17). Culture supernatants from the S. typhimurium strain SB221 prepared by precipitation with trichloroacetic acid and acetone as previously described (18) were separated on SDS/polyacrylamide gels and transferred to poly(vinylidene difluoride) (PVDF) membranes (Fig. 1). Blotted proteins were either sequenced directly by automated Edman degradation or digested with trypsin to generate internal peptide fragments. Peptide fragments were separated by reversed-phase HPLC and sequenced as described (23). The N-terminal sequence (Mq/nIQSFYaAKLKTQ) (lowercase letters indicate ambiguous determination) of the 62-kDa protein (Fig. 1) showed no significant similarity to any proteins in the available databases. However, an internal fragment of this protein (VLLNSGNLEIQK) showed significant homology to IpgD, a protein of similar size from Shigella spp. with no identified function that is secreted by means of a plasmid-encoded type III secretion system (24). When these experiments were performed, no sequences with homology to either the N-terminal (TKITLSPQNFRIQKQETTLLKEK) or any of the internal peptide sequences (NTESSATHFHR; EAILSAVYSK; NHFIELR; NDVFTPSGAGANPFI) from the 25-kDa protein (Fig. 1) were identified in the available databases (however, see below). Antibodies to culture supernatant proteins electroeluted from SDS/10% polyacrylamide gels were prepared as described elsewhere (18).
Figure 1.
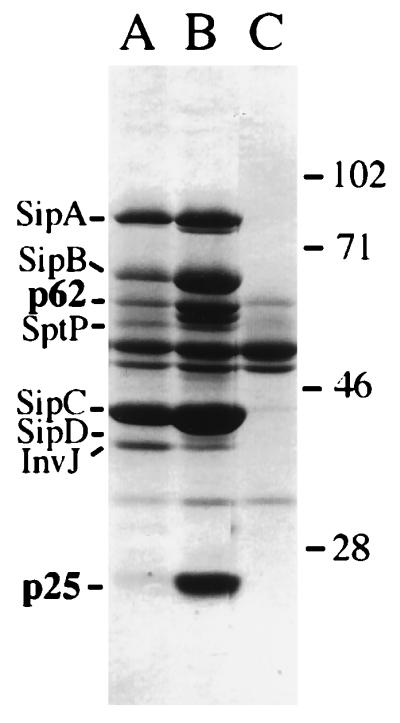
Type III secreted proteins of S. typhimurium. Culture supernatant proteins of wild-type S. typhimurium (lane A) and the isogenic sipD (lane B) and invG (lane C) strains were separated on an SDS/10% polyacrylamide gel and stained with Coomassie blue.
Recombinant DNA, Genetic Techniques, and Nucleotide Sequencing.
Construction and screening of a λgt11 expression library of S. typhimurium SL1344 total-cell DNA and was carried out as described elsewhere (25). DNA inserts of recombinant clones that reacted with the antibody (6 recombinant bacteriophage clones of ≈300,000 screened) were subcloned into pBluescript SK II (+) as KpnI/SacI fragments and their nucleotide sequences were determined by the dideoxynucleotide chain-termination method. By comparison with the peptide sequences of the 25-kDa protein, we identified a plasmid (pSB898) with a 1.2-kb insert from a recombinant λgt11 clone that carried the complete gene for this protein (sopE).
Chromosomal DNA adjacent to sopE was retrieved by chromosomal walking as described elsewhere (17) and its nucleotide sequence was determined by the dideoxynucleotide chain-termination method.
Strains carrying nonpolar mutations in orfJ (SB756) or sopE (SB856) were constructed by allele exchange as described elsewhere (15). SB757, a S. typhimurium strain carrying a polar mutation in sopE, was constructed by chromosomal integration of a derivative of the suicide vector pGP704 (pSB1128, ampicillin-resistant) carrying an internal fragment (nucleotides 40–311) of sopE.
An S. typhimurium strain expressing a SopE protein fused to an epitope recognized by the monoclonal antibody M45 and consisting of 18 residues from the E4-6/7 protein of adenovirus (MDRSRDRLPPFETETRIL) (26) was constructed by PCR using the epitope-tagging vector pSB504 (20). The epitope-tagged sopE allele was integrated by single recombination into the chromosome of the wild-type strain SL1344 by allelic exchange as described above, yielding strain SB875. Derivatives of this strain carrying mutations in invC (SB882), invG (SB883), and sipD (SB884) were constructed by P22-mediated transduction. Hfr-mediated mapping was performed essentially as described by Sanderson and Demerec (27). P22HTint-mediated transduction was carried out as described elsewhere (28).
Virulence Assays, Immunofluorescence Staining, and Analysis of Cytoskeletal Rearrangements in Infected Host Cells.
Bacterial internalization into cultured Henle-407 epithelial cells by the gentamicin-resistance assay (29), J774 macrophage cytotoxicity assays (22), and experimental animal infections (29) were carried out as described elsewhere. Analyses of cytoskeletal rearrangements by rhodamine-labeled phalloidin staining and bacterial localization in infected cells by immunofluorescence staining were carried out as previously described (30). The ability of S. typhimurium strains to rescue the invasion phenotype of the noninvasive strain SB169 was measured as previously described (9). Briefly, cultured Henle-407 cells were infected with equal numbers of either wild-type or sopE S. typhimurium strains (both kanamycin sensitive) and the noninvasive sipB strain SB169 (kanamycin resistant) at various multiplicities of infection (moi = 0.3–5). The number of internalized bacteria after 30 or 60 min of infection was measured by the gentamicin-resistance assay. The number of internalized S. typhimurium SB169 (kanamycin resistant) was specifically determined by plating bacteria in the presence of kanamycin.
RESULTS
Identification of S. typhimurium SopE Protein.
S. typhimurium relies on the function of the centisome 63 type III protein secretion system to modulate host cellular functions (3). Characterization of proteins that are secreted by this system is of great interest, as it may lead to the identification of potential effectors of cellular responses. Although several of these secreted proteins have been identified (5, 7, 17–19, 21, 31), a number of polypeptides secreted into the culture supernatant by means of the centisome 63 type III system remained uncharacterized. In addition, it is unknown which, if any, of these uncharacterized polypeptides detected in Coomassie blue-stained SDS/polyacrylamide gels of S. typhimurium culture supernatants represent degradation products of larger secreted proteins. We identified a 25-kDa secreted protein of S. typhimurium and cloned its coding gene as indicated in Materials and Methods. The cloned gene was found to encode a 725-nt open reading frame (ORF) that exactly matched the amino acid sequences from the various fragments of the 25-kDa protein (see Materials and Methods). In addition, culture supernatants from an S. typhimurium strain carrying a nonpolar null mutation in this ORF (see Materials and Methods) lacked the 25-kDa protein (Fig. 2). The presence of the 25-kDa protein in the culture supernatant of this strain was restored by the introduction of a plasmid carrying the 725-nt ORF. These results indicate that the cloned 725-nt ORF encodes the 25-kDa secreted protein. During the course of the characterization of the 25-kDa S. typhimurium secreted protein, Wood et al. (5) published the sequence of an S. dublin gene, termed sopE, which is 90% identical to the ORF encoding the p25 protein. Because it is likely that the p25 protein is the homolog of SopE, we will refer to the S. typhimurium protein as SopE.
Figure 2.

Western blot analysis of SopE secretion. (I) Effect of targets and components of the centisome 63 type III secretion system on SopE secretion. Proteins from culture supernatants of the wild-type S. typhimurium SL1344 and the isogenic strains with nonpolar mutations in sopE (SB757), invG (SB161), sipA (SB225), sipB (SB169), sipC (SB220), sipD (SB221), sptP (SB237), spaO (SB302), or invJ (SB303), as well as from SB757 complemented with pSB1130 (which carries sopE) were resolved on an SDS/12% polyacrylamide gel, transferred to a nitrocellulose membrane, and sequentially probed with antibodies directed against SopE and SipB. Reprobing for the intracellular marker protein 6-phosphogluconate dehydrogenase verified that the observed pattern of supernatant proteins was not due to bacterial lysis (data not shown). (II) Secretion of epitope-tagged SopE. Culture supernatant proteins and whole-cell lysates (duplicate independent samples, a and b) from a wild-type S. typhimurium strain expressing, under the control of its native promoter, a chromosomally encoded, M45-epitope-tagged SopE (SB875), and its isogenic derivatives carrying mutations in invG (SB882), invC (SB883), or sipD (SB884), were separated on an SDS/12% polyacrylamide gel, transferred to nitrocellulose, and probed with a mouse monoclonal antibody directed to the epitope tag.
SopE Is a Target of the Centisome 63 Type III Protein Secretion System.
To confirm that SopE is secreted by means of the centisome 63 type III secretion system, we performed immunoblot analyses of culture supernatants from wild-type S. typhimurium, a derivative strain encoding an M45-epitope-tagged SopE, and several isogenic mutants. SopE was absent from the supernatants of strains carrying mutations in invG (15), spaO (20), or invJ (21), which are essential for secretion by the centisome 63 type III secretion system (Fig. 2I). The absence of SopE from the culture supernatant of these strains was not due to a defect in sopE expression, as an epitope-tagged version of SopE was readily detectable in whole cell lysates of secretion-defective mutant strains (Fig. 2II). SopE was detected in culture supernatants of strains carrying mutations in genes encoding the secreted proteins SipA (17), SipC (18), and SptP (19) (Fig. 2I). Strains carrying mutations in the genes encoding the secreted proteins SipB or SipD, which have been previously shown to exhibit increased secretion by the centisome 63 type III system (17), also exhibited increased levels of SopE in their culture supernatants (Fig. 2I). These results confirm that SopE is indeed a target of the centisome 63 type III secretion system. Inactivation of sopE had no effect on the secretion of the other known targets of this system (Fig. 2I) or on the translocation of SipC into the host cell (data not shown). These data indicate that SopE is not involved in either secretion or translocation through the type III secretion system, and they suggest that SopE may be an effector protein that exerts its function inside the eukaryotic host cell. This is consistent with the observation that SopE from S. typhimurium (W.-D.H. and J.E.G., unpublished results) and from S. dublin is translocated into host cells (5).
Phenotype of S. typhimurium sopE Mutants.
To analyze the role of SopE in S. typhimurium pathogenesis, we compared the virulence of wild-type S. typhimurium SL1344 and the isogenic sopE mutant strains SB757 and SB856 in several in vivo and in vitro assays. Inactivation of sopE had no effect on either the LD50 or the mean time to death of orally inoculated BALB/c mice (Table 1). Similarly, the introduction of a sopE mutation did not affect the macrophage cytotoxicity of S. typhimurium (Table 1).
Table 1.
Phenotypes of S. typhimurium sopE mutant
| Strain | Relevant genotype | Henle-407 invasion,* % | J774 toxicity,† % | Mouse virulence (mean time to death),‡ days | Rescue of invasion phenotype,§ % |
|---|---|---|---|---|---|
| SL1344 | Wild type | 42 ± 8 | 23 ± 5 | 6 | 100 ± 10 |
| SB136 | invA::aphT | 0.2 ± 0.05 | 3 ± 1 | ND | <1 |
| SB757 | sopE | 40 ± 5 | 21 ± 5 | 6 | 40 ± 5 |
| SB757 (pSB1130) | sopE (sopE+) | 53 ± 10 | ND | ND | 100 ± 10 |
| SB756 | orfJ:aphT | 43 ± 5 | 24 ± 5 | 6 | ND |
Entry into Henle-407 cells was determined by the gentamicin-resistance assay. Values are the mean ± the standard deviation of six independent determinations and represent the percentage of the bacterial inoculum that survived 2 h of gentamicin treatment. Equivalent results were obtained with strain SB856.
Macrophage J774 cytotoxicity was determined by a dye exclusion assay (22). Values are the percentage of cells exhibiting cytotoxicity after 30 min of infection. At least 200 cells were evaluated in each assay. Equivalent results were obtained with strain SB856. ND, not done.
BALB/c mice (six per group) were infected orally with 106 bacteria. Virulence is given as mean time to death. There were no survivors in any group.
The ability of the different strains to rescue the entry phenotype of the invasion-defective S. typhimurium sipB strain SB169 was measured as described in the text. Values were standardized considering the rescue ability of wild type to be 100%. Equivalent results were obtained with strain SB856.
When measured by the gentamicin-resistance assay, the sopE mutant exhibited levels of entry into cultured Henle-407 cells that were indistinguishable from those of the wild-type strain (Table 1). However, during the course of these experiments we observed that the morphology of the membrane ruffles required for bacterial entry induced by the sopE mutant was different from that of the ruffles induced by the wild-type strain. Rhodamine-phalloidin staining of cells infected with the sopE strain SB757 revealed that the actin cytoskeletal rearrangements induced by this strain were less extensive than those induced by wild type (Fig. 3I). This difference was particularly noticeable at earlier time points (15 min) after infection (Fig. 3I). To examine whether the altered cytoskeletal rearrangements induced by the sopE strain translate into a defect in cell invasion, we employed an assay based on differential staining of internalized bacteria that allowed us to measure entry levels after short infection times (30). When measured by this assay, the presence of the sopE mutation caused a 1.6-fold decrease in the internalization levels after 20 min of infection relative to the wild-type strain (Fig. 3II).
Figure 3.

Effect of a null mutation in sopE on the interaction of S. typhimurium with Henle-407 cells. (I) Cultured Henle-407 cells were infected with S. typhimurium for 15 min, fixed, and stained with rhodamine-labeled phalloidin to visualize the actin cytoskeleton (A) and with a fluorescein isothiocyanate (FITC)-labeled antibody directed to S. typhimurium to visualize bacteria (B). (C) Phase-contrast images of the same cells. (Micrographs obtained with a ×40 objective.) (II) Internalization of the S. typhimurium sopE mutant into Henle-407 cells. Bacterial internalization levels, measured by microscopy as indicated in the text, were standardized to wild-type S. typhimurium. At a minimum, 300 cells were scored for each strain. The standard deviation of these experiments was less than 10%. The strains were as follows: wild type, SL1344; sopE, SB757; sopE (psopE), SB757 (pSB1130); invG, SB161.
It has previously been shown that wild-type S. typhimurium can rescue the entry phenotype of invasion-deficient strains when simultaneously applied to cultured epithelial cells (9, 10). We tested the ability of the S. typhimurium sopE mutant to rescue the invasion phenotype of the noninvasive sipB mutant strain SB169. We reasoned that the reduced ability of the sopE mutant to induce membrane ruffling may result in a reduced ability to rescue the entry of an invasion-defective mutant. Indeed, the sopE mutant had a 2.5 ± 0.3-fold lower ability, relative to the wild-type strain SL1344, to rescue the invasion phenotype of the sipB mutant (Table 1). This defect was observed over a range of multiplicities of infection and was complemented by supplying the sopE gene in trans (Table 1). Together, these results indicate that SopE is involved in promoting efficient S. typhimurium entry into nonphagocytic cells, possibly by enhancing the bacterial-induced cytoskeletal rearrangements.
Chromosomal Mapping of the sopE Locus.
All proteins secreted by means of type III secretion systems characterized to date are encoded in virulence-associated plasmids or pathogenicity islands in the immediate vicinity of the genes that encode the actual secretion apparatus (reviewed in ref. 4). Because SopE is a target of the type III secretion system encoded in the centisome 63 pathogenicity island, we investigated whether it was encoded in this pathogenicity island. Southern hybridization and P22-mediated linkage analyses using different markers located throughout the centisome 63 region indicated that sopE was not encoded in this pathogenicity island of S. typhimurium (data not shown). We then determined the location of sopE in the Salmonella chromosome by Hfr-mediated marker-linkage analysis. An aphT insertion located immediately adjacent to sopE was transferred into the chromosome of several S. typhimurium Hfr strains with different origins of transfer. An Hfr strain with a clockwise origin of transfer at centisome 43 was shown to transfer the aphT insertion at high frequencies after short mating times. Further Hfr-mediated mapping and marker co-transfer experiments established that sopE is located at centisome 61 of the S. typhimurium chromosome (data not shown).
Genetic Organization of the sopE Chromosomal Region.
To study the sopE region in more detail, we cloned the chromosomal region of wild-type S. typhimurium strain SL1344 that is located immediately adjacent to this gene. DNA fragments of 1.2 kb and 3.7 kb located immediately upstream and downstream of sopE, respectively, were retrieved by chromosomal walking (see Materials and Methods) and their entire nucleotide sequence was determined. DNA sequence analyses revealed the presence of several ORFs that show significant sequence similarity to tail proteins from several bacteriophages and invertible genetic elements (Fig. 4). Immediately downstream of sopE, we identified two ORFs that exhibited sequence similarity to the tail fiber proteins OrfK and Orf45 of bacteriophage 186 (Fig. 4). Interestingly, the identified ORFs encode proteins that exhibit a “mosaic” structure, with discrete regions of homology to several otherwise unrelated bacteriophages such as P1 (32), P2 (33), 186 (34), and T2 and K3 (35), as well as tail fiber-like proteins encoded in invertible elements of the Shigella chromosome (36). This chimeric structure is characteristic of proteins that determine the host range of bacteriophages and is thought to have arisen through illegitimate recombination between tail fiber genes from otherwise unrelated bacteriophages (37). Four other ORFs exhibiting sequence similarity to lambdoid bacteriophage genes were identified upstream of sopE. These ORFs encode putative proteins with strong amino acid sequence similarity over their entire length to the tail-sheath proteins OrfJ, OrfI, OrfH, and OrfG of bacteriophage 186 (34) as well as proteins FI and FII from phage P2 (38) and to Orf12 of a cryptic P2-like phage from Haemophilus somnus (39) (Fig. 4). Overall, the sequence similarity is particularly striking when compared with proteins from the P2-like phage 186. In this case, the conservation extends to the genetic organization of the homologous genes (Fig. 4). Interestingly, the attachment site for bacteriophage 186 integration in the E. coli chromosome is located between pheA and nalB (40), which is similar to the location of the cryptic bacteriophage that encodes sopE in S. typhimurium strain SL1344.
Figure 4.

Comparison of the genetic organization of the S. typhimurium sopE chromosomal region with that of P2-like phages. Numbers in circles denote the percent identity between the indicated protein sequences.
Immediately upstream of sopE, there is a 212-nt noncoding region followed by an ORF (orfR) encoding a putative protein of 68 amino acids with a high degree of similarity to the C-terminal helix–turn–helix DNA-binding domain of the resolvase family of site-specific recombinases (Fig. 5A). This family of proteins includes the DNA invertases encoded by pin, min, and hin from Escherichia coli and S. typhimurium (41, 42), as well as the DNA invertases from bacteriophages Mu, P1, and P7 (43, 44). The Salmonella OrfR lacks the N-terminal catalytic domain characteristic of these proteins, which harbors a conserved serine residue that is transiently attached to the DNA during catalysis (45), and there is no identifiable Shine-Dalgarno sequence near the putative OrfR start codon, suggesting that this ORF may not be functional and might have been disrupted by illegitimate recombination. However, 1,293 nt downstream of sopE, there is a 26-nt stretch that fits the consensus target site for this family of site-specific recombinases (46) (Fig. 5B).
Figure 5.

(A) Sequence alignment of S. typhimurium OrfR and site-specific recombinases. (B) Sequence alignment of a putative binding site for site-specific recombinases located 1,293 nt downstream of sopE, with the consensus binding sequence for this protein family (46).
Distribution of sopE in Various S. enterica Serovars.
The distribution of sopE among different Salmonella enterica serovars was investigated by dot-blot analyses of chromosomal DNA with an internal fragment of sopE as a probe. The presence of sopE was detected in only a limited subset of S. enterica serovars, including S. typhi, S. heidelberg, S. hadar, S. newport, S. dublin, S. enteritidis, and S. pullorum (Table 2). No pattern emerged correlating the presence of sopE with either serotype or host specificity. In fact, sopE was not detected in some isolates derived from the LT2 strain of S. typhimurium (Fig. 6). Southern hybridization analyses of a selected group of strains revealed a high degree of restriction fragment polymorphism in the sopE region of the Salmonella chromosome, indicating that this gene is located in a region that undergoes frequent recombination (Fig. 6).
Table 2.
Dot-blot hybridization to assess distribution of sopE
| Serovar | Serotype |
|---|---|
| Positive hybridization signal | |
| S. enterica sv. heidelberg | B |
| S. enterica sv. typhimurium (SL1344, SR11, 68500) | B |
| S. enterica sv. hadar | C2 |
| S. enterica sv. newport | C2 |
| S. enterica sv. dublin | D1 |
| S. enterica sv. enteritidis | D1 |
| S. enterica sv. pullorum (#3045, #3648) | D1 |
| S. enterica sv. typhi (TY2, ISP1820, ISP2822) | D1 |
| Negative hybridization signal | |
| S. enterica sv. typhimurium (LT2 derivatives χ3364, SA535) | B |
| S. enterica sv. agona | B |
| S. enterica sv. brandenburg | B |
| S. enterica sv. bredeney | B |
| S. enterica sv. duisburg | B |
| S. enterica sv. java | B |
| S. enterica sv. schwarzengrund | B |
| S. enterica sv. braenderup | C1 |
| S. enterica sv. choleraesuis | C1 |
| S. enterica sv. infantis | C1 |
| S. enterica sv. montevideo | C1 |
| S. enterica sv. ohio | C1 |
| S. enterica sv. othmarschen | C1 |
| S. enterica sv. tennessee | C1 |
| S. enterica sv. thompson | C1 |
| S. enterica sv. virchov | C1 |
| S. enterica sv. bovis morbidificans | C2 |
| S. enterica sv. manhattan | C2 |
| S. enterica sv. nienstaedten | C4 |
| S. enterica sv. panama | D1 |
| S. enterica sv. anatum | E1 |
| S. arizona | – |
When more than one isolate was tested, strain names are given in parentheses.
Figure 6.

Southern hybridization analyses of the sopE and orfJ regions of Salmonella spp. Total cell DNA from various S. enterica serovars, digested with HindIII, EcoRV, or both, was probed with a 0.7-kb HindIII/XmnI fragment comprising the 3′ portion of orfJ and the 5′ portion of orfI. Blots were subsequently stripped and reprobed with a DNA fragment corresponding to the coding region of sopE.
The presence of phage-homologous sequences was also investigated by dot-blot hybridization using different probes derived from phage sequences located either upstream (orfJ and orfG) or downstream (orfK) of sopE. Although these probes hybridized to DNA of most serovars analyzed, the intensity of the signal varied widely, indicating wide distribution and a great deal of sequence divergence in this family of apparently related phages (data not shown). Southern hybridization analysis of a discrete set of strains with a probe containing phage sequences revealed significant restriction fragment length polymorphism (Fig. 6). Interestingly, some serovars of S. enterica (e.g., S. gallinarum) contain sequences that hybridize to the sopE probe but lack sequences that hybridize to the phage probe. Conversely, some serovars of S. enterica (e.g., S. java and some isolates of S. typhimurium) hybridized to the phage probe but did not hybridize to the sopE probe (Fig. 6).
DISCUSSION
The centisome 63 type III secretion system of S. typhimurium plays a central role in the interaction of these bacteria with host cells (3). A number of polypeptides that are secreted via this pathway have been detected in SDS/PAGE gels (5, 17, 18, 31). Although the identity of several of these proteins has been established (5, 7, 17–19, 21, 31), a number of these polypeptides remain uncharacterized. We report here the identification of one of these secreted proteins, which is the S. typhimurium homolog of the recently reported SopE protein from S. dublin (5). We have demonstrated that S. typhimurium SopE is secreted into the supernatants of S. typhimurium cultures by means of the dedicated type III protein secretion system encoded at centisome 63 of the Salmonella chromosome. An S. typhimurium strain carrying a null mutation in sopE was deficient in its ability to gain access to cultured cells after short infection times. Furthermore, the S. typhimurium sopE mutant induced membrane ruffles that appear to be more diffuse than those induced by the wild-type strain. Consistent with this observation, a sopE mutant strain was less efficient than the wild-type strain at rescuing entry of an invasion-defective sipB mutant strain in a co-infection experiment. The S. typhimurium sopE mutant strain was able to secrete and/or translocate into host cells other targets of the centisome 63 type III secretion system at levels equivalent to the wild-type strain. This suggests that SopE is not involved in either secretion or translocation and may, therefore, function as an effector of cellular responses. Consistent with this hypothesis, SopE from S. typhimurium (W.-D.H. and J.E.G., unpublished results) and from S. dublin (5) have been shown to be translocated into host cells. When longer infection times were used in the experimental assays, the S. typhimurium sopE strain was able to enter cultured Henle-407 cells at levels that were virtually indistinguishable from those of the wild-type strain. Thus, although SopE does play a role in modulating efficient bacterial entry, S. typhimurium must encode other determinants with redundant functions. This conclusion is supported by our observation that sopE does not impair the virulence of S. typhimurium in a mouse model of oral infection.
We have demonstrated that sopE is not linked to the centisome 63 pathogenicity island, where the genes encoding the type III secretion apparatus that direct its secretion and translocation are located (47). This finding was surprising because all previously identified targets of type III secretion systems of animal or plant pathogenic bacteria have been shown to be encoded in the immediate vicinity of the genes encoding the secretion apparatus (reviewed in ref. 4). Molecular genetic analyses revealed that sopE is encoded within the genome of a cryptic bacteriophage located at centisome 61 of the S. typhimurium chromosome. Nucleotide sequence analysis of the region bordering sopE revealed the presence of several ORFs with strong sequence similarity to tail proteins of several lambdoid bacteriophages. The similarity to the P2-like bacteriophage 186 extended to the genetic organization of the homologous genes. Interestingly, the integration site for bacteriophage 186 in the E. coli chromosome has been mapped to a region equivalent to the location of sopE in the S. typhimurium chromosome (40). We were unsuccessful in detecting lytic induction of the prophage with procedures that have been reported to result in the induction of related prophages (data not shown) (48). We also failed to detect transfer of antibiotic-resistance markers placed in either sopE or neighboring phage genes to recipient strains of S. typhimurium. Therefore, it is likely that the sopE-bearing bacteriophage is defective. The presence of defective bacteriophages in the genomes of Gram-negative bacteria is a relatively common occurrence (37). Alternatively, an as-yet-unknown inducing signal, perhaps derived from the host, may be required for phage induction. Indeed, several examples of phage induction and modulation of horizontal gene transfer by host signals have been reported (49, 50). If indeed the sopE-encoding phage is defective, it is an intriguing possibility that this phage may have been initially proficient and that the potential selective advantage of encoding sopE may have selected for a defective phage as a mechanism to stabilize this trait.
Hybridization analyses revealed that sopE is present in only a small subset of Salmonella serotypes. This is in marked contrast to genes encoding the type III secretion machinery that directs the secretion and translocation of SopE. These genes are widely distributed among all Salmonella serotypes (51, 52). Restriction fragment length polymorphism analyses of chromosomal DNA from different Salmonella serovars by using orfJ (Fig. 6) or orfK (data not shown) as probes revealed a great deal of heterogeneity in the bacteriophage-like sequences present in these strains. Interestingly, some strains of Salmonella showed either no or weak hybridization to orfJ, further demonstrating the variable nature of these sequences in the Salmonella chromosome.
Our findings suggest that sopE may have been acquired recently by S. typhimurium as a consequence of the integration of a lysogenic phage. Our inability to demonstrate bacteriophage induction suggests that this bacteriophage may be defective. However, even if the cryptic bacteriophage of this isolate of S. typhimurium is defective, the location of sopE flanked by bacteriophage genes that are known to undergo frequent recombination may eventually allow this gene to associate with a proficient bacteriophage. This, in turn, would significantly increase the potential for the horizontal transfer of sopE to other strains. The widespread presence of generalized transducing phages in natural isolates of Salmonella may also contribute to increase the likelihood of this event. Indeed, a recent study showed that of 174 natural isolates of Salmonella spp. tested, more than 90% were lysogenic for P22-like phages and more than 95% of those were able to transduce chromosomal and plasmid markers (53, 54).
It is now evident that the centisome 63 type III secretion system is involved in a variety of pathogenic mechanisms in addition to bacterial entry. In addition, we show here that the array of secreted target proteins that travel through this system differs among Salmonella serotypes. It is possible that in some cases the acquisition or loss of type III secreted proteins may be associated with adaptation to a new host. In this context, the presence of secreted effector target proteins such as SopE encoded in a bacteriophage may confer additional advantage for the rapid adaptation of Salmonella species to a new environment or a new host. High horizontal mobility of effector protein genes may therefore allow the bacteria to maximize the chances of assembling the most appropriate set of effector molecules for a given host or environment while at the same time maintaining the genes required for the secretion and translocation of these proteins.
Acknowledgments
We thank Manuel Cruz and Sumati Murli for critical review of this manuscript. This work was supported by Public Health Service Grants AI30492 and GM52543 from the National Institutes of Health to J.E.G., who is an investigator of the American Heart Association. W.-D.H. was supported in part by a fellowship from the German Bundesministerium für Forschung und Technologie.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF043239).
References
- 1.Ochman H, Soncini F C, Solomon F, Groisman E A. Proc Natl Acad Sc USA. 1996;93:7800–7804. doi: 10.1073/pnas.93.15.7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shea J E, Hensel M, Gleeson C, Holden D W. Proc Natl Acad Sci USA. 1996;93:2593–2597. doi: 10.1073/pnas.93.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galán J E. Mol Microbiol. 1996;20:263–271. doi: 10.1111/j.1365-2958.1996.tb02615.x. [DOI] [PubMed] [Google Scholar]
- 4.Galán J E, Bliska J B. Annu Rev Cell Dev Biol. 1996;12:219–253. doi: 10.1146/annurev.cellbio.12.1.221. [DOI] [PubMed] [Google Scholar]
- 5.Wood M W, Rosqvist R, Mullan P B, Edwards M H, Galyov E E. Mol Microbiol. 1996;22:327–338. doi: 10.1046/j.1365-2958.1996.00116.x. [DOI] [PubMed] [Google Scholar]
- 6.Collazo C, Galán J E. Mol Microbiol. 1997;24:747–756. doi: 10.1046/j.1365-2958.1997.3781740.x. [DOI] [PubMed] [Google Scholar]
- 7.Hardt W-D, Galán J E. Proc Natl Acad Sci USA. 1997;94:9887–9892. doi: 10.1073/pnas.94.18.9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groisman E A, Ochman H. Cell. 1996;87:791–794. doi: 10.1016/s0092-8674(00)81985-6. [DOI] [PubMed] [Google Scholar]
- 9.Ginocchio C, Pace J, Galán J E. Proc Natl Acad Sci USA. 1992;89:5976–5980. doi: 10.1073/pnas.89.13.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galán J E, Ginocchio C, Costeas P. J Bacteriol. 1992;17:4338–4349. doi: 10.1128/jb.174.13.4338-4349.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groisman E A, Ochman H. EMBO J. 1993;12:3779–3787. doi: 10.1002/j.1460-2075.1993.tb06056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altmeyer R M, McNern J K, Bossio J C, Rosenshine I, Finlay B B, Galán J E. Mol Microbiol. 1993;7:89–98. doi: 10.1111/j.1365-2958.1993.tb01100.x. [DOI] [PubMed] [Google Scholar]
- 13.Ginocchio C, Rahn K, Clarke R C, Galán J E. Infect Immun. 1997;65:1267–1272. doi: 10.1128/iai.65.4.1267-1272.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoiseth S K, Stocker B A. Nature (London) 1981;291:238–239. doi: 10.1038/291238a0. [DOI] [PubMed] [Google Scholar]
- 15.Kaniga K, Bossio J C, Galán J E. Mol Microbiol. 1994;13:555–568. doi: 10.1111/j.1365-2958.1994.tb00450.x. [DOI] [PubMed] [Google Scholar]
- 16.Eichelberg K, Ginocchio C, Galán J E. J Bacteriol. 1994;176:4501–4510. doi: 10.1128/jb.176.15.4501-4510.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaniga K, Trollinger D, Galán J E. J Bacteriol. 1995;177:7078–7085. doi: 10.1128/jb.177.24.7078-7085.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaniga K, Tucker S C, Trollinger D, Galán J E. J Bacteriol. 1995;177:3965–3971. doi: 10.1128/jb.177.14.3965-3971.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaniga K, Uralil J, Bliska J B, Galán J E. Mol Microbiol. 1996;21:633–641. doi: 10.1111/j.1365-2958.1996.tb02571.x. [DOI] [PubMed] [Google Scholar]
- 20.Collazo C, Galán J E. Infect Immun. 1996;64:3524–3531. doi: 10.1128/iai.64.9.3524-3531.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collazo C M, Zierler M K, Galán J E. Mol Microbiol. 1995;15:25–38. doi: 10.1111/j.1365-2958.1995.tb02218.x. [DOI] [PubMed] [Google Scholar]
- 22.Chen L M, Kaniga K, Galán J E. Mol Microbiol. 1996;21:1101–1115. doi: 10.1046/j.1365-2958.1996.471410.x. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez J, deMott M, Atherton D, Mische S M. Anal Biochem. 1992;201:255–264. doi: 10.1016/0003-2697(92)90336-6. [DOI] [PubMed] [Google Scholar]
- 24.Hueck C J, Hantman M J, Bajaj V, Johnston C, Lee C A, Miller S I. Mol Microbiol. 1995;18:479–490. doi: 10.1111/j.1365-2958.1995.mmi_18030479.x. [DOI] [PubMed] [Google Scholar]
- 25.Allaoui A, Menard R, Sansonetti P J, Parsot C. Infect Immun. 1993;61:1707–1714. doi: 10.1128/iai.61.5.1707-1714.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 27.Obert S, O’Connor R J, Schmid S, Hearing P. Mol Cell Biol. 1994;14:1333–1346. doi: 10.1128/mcb.14.2.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanderson K E, Demerec M. Genetics. 1965;51:897–913. doi: 10.1093/genetics/51.6.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmieger H. Mol Gen Genet. 1972;119:74–88. doi: 10.1007/BF00270447. [DOI] [PubMed] [Google Scholar]
- 30.Galán J E, Curtiss R., III Proc Natl Acad Sci USA. 1989;86:6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L M, Hobbie S, Galán J E. Science. 1996;274:2115–2118. doi: 10.1126/science.274.5295.2115. [DOI] [PubMed] [Google Scholar]
- 32.Sandmeier H, Iida S, Arber W. J Bacteriol. 1992;174:3936–3944. doi: 10.1128/jb.174.12.3936-3944.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haggard-Ljungquist E, Halling C, Calendar R. J Bacteriol. 1992;174:1462–1477. doi: 10.1128/jb.174.5.1462-1477.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xue Q, Egan J B Virology. Virology. 1995;212:128–133. doi: 10.1006/viro.1995.1461. [DOI] [PubMed] [Google Scholar]
- 35.Riede I, Drexler K, Eschbach M L, Henning U. J Mol Biol. 1986;191:255–266. doi: 10.1016/0022-2836(86)90262-7. [DOI] [PubMed] [Google Scholar]
- 36.Tominaga A, Ikemizu S, Enomoto M. J Bacteriol. 1991;173:4079–4087. doi: 10.1128/jb.173.13.4079-4087.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell A. In: Phage Evolution and Speciation. Calendar R, editor. Vol. 1. New York: Plenum; 1988. pp. 1–17. [Google Scholar]
- 38.Temple L M, Forsburg S L, Calendar R, Christie G E. Virology. 1991;181:353–358. doi: 10.1016/0042-6822(91)90502-3. [DOI] [PubMed] [Google Scholar]
- 39.Pontarollo R A, Rioux C R, Potter A A. J Bacteriol. 1997;179:1872–1879. doi: 10.1128/jb.179.6.1872-1879.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woods W H, Egan J B. J Virol. 1974;14:1349–1354. doi: 10.1128/jvi.14.6.1349-1356.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plasterk R H, Brinkman A, van de Putte P. Proc Natl Acad Sci USA. 1983;80:5355–5358. doi: 10.1073/pnas.80.17.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zieg J, Simon M. Proc Natl Acad Sci USA. 1980;77:4196–4200. doi: 10.1073/pnas.77.7.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ritthaler W, Kamp D. Nucleic Acids Res. 1988;16:6246. doi: 10.1093/nar/16.13.6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klippel A, Cloppenborg K, Kahmann R. EMBO J. 1988;7:3983–3989. doi: 10.1002/j.1460-2075.1988.tb03286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garnier T, Saurin W. Mol Microbiol. 1987;1:371–376. doi: 10.1111/j.1365-2958.1987.tb01944.x. [DOI] [PubMed] [Google Scholar]
- 46.Sandmeier H. Mol Microbiol. 1994;12:343–350. doi: 10.1111/j.1365-2958.1994.tb01023.x. [DOI] [PubMed] [Google Scholar]
- 47.Mills D B, Bajaj V, Lee C A. Mol Microbiol. 1995;15:749–759. doi: 10.1111/j.1365-2958.1995.tb02382.x. [DOI] [PubMed] [Google Scholar]
- 48.Bradley C, Ling O P, Egan J B. Mol Gen Genet. 1975;140:123–135. doi: 10.1007/BF00329780. [DOI] [PubMed] [Google Scholar]
- 49.Mahan M J, Slauch J M, Mekalanos J J. In: Environmental Regulation of Virulence Gene Expression in Escherichia coli, Salmonella, and Shigella spp. Neidhardt F N, editor. Vol. 2. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 2803–2815. [Google Scholar]
- 50.Waldor M K, Mekalanos J J. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 51.Galán J E, Curtiss R., III Infect Immun. 1991;59:2901–2908. doi: 10.1128/iai.59.9.2901-2908.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rahn K, De Grandis S, Clarke R C, McEwen S A, Galán J E, Ginocchio C, Curtiss III R, Gyles C L. Mol Cell Probes. 1992;6:271–279. doi: 10.1016/0890-8508(92)90002-f. [DOI] [PubMed] [Google Scholar]
- 53.Schicklmaier P, Schmieger H. Appl Env Microbiol. 1995;61:1637–1640. doi: 10.1128/aem.61.4.1637-1640.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmieger H, Schicklmaier P, Zimmer A. In: Salmonella Phages: Frequency, Transduction, and Use of Their Genome Restriction Patterns as Markers for Host Typing. Colin P, Le Goux J M, Clement G, editors. Ploufragan, France: Zoopole; 1997. pp. 23–27. [Google Scholar]