

Abstract
In multiple myeloma, karyotypic 14q32 translocations have been identified at a variable frequency (10–60% in different studies). In the majority of cases, the partner chromosome has not been identified (14q+), and in the remaining cases, a diverse array of chromosomal partners has been implicated, with 11q13 being the most common. We developed a comprehensive Southern blot assay to identify and distinguish different kinds of immunoglobulin heavy chain (IgH) switch recombination events. Illegitimate switch recombination fragments (defined as containing sequences from only one switch region) are potential markers of translocation events into IgH switch regions and were identified in 15 of 21 myeloma cell lines, including seven of eight karyotyped lines that have no detectable 14q32 translocation. From all nine lines or tumor samples analyzed further, cloned illegitimate switch recombination fragments were confirmed to be IgH switch translocation breakpoints. In three of these cases, the translocation breakpoint was shown to be present in the primary tumor. These translocation breakpoints involve six chromosomal loci: 4p16.3 (two lines and the one tumor); 6; 8q24.13; 11q13.3 (in three lines); 16q23.1; and 21q22.1. We suggest that translocations into the IgH locus (i) are frequent (karyotypic 14q32 translocations and/or illegitimate switch recombination fragments are present in primary tumor samples and in 19 of 21 lines that we have analyzed); (ii) occur mainly in switch regions; and (iii) involve a diverse but nonrandom array (i.e., frequently 11q13 or 4p16) of chromosomal partners. This appears to be the most frequent genetic abnormality in multiple myeloma.
Keywords: chromosome translocation, isotype switch recombination, cyclin D1, immunoglobulin heavy chain gene
Multiple myeloma (MM) is a uniformly fatal malignancy that accounts for about 10% of all hematopoietic malignancies, and often arises from a common premalignant condition called monoclonal gammopathy of undetermined significance (MGUS) that is present in 1% of the adult population (1, 2). In MM, different studies indicate that translocations involving band 14q32 [site of the immunoglobulin heavy chain (IgH) locus] occur at a frequency of 10–60%. However, with the exception of involvement of 11q13 in about 30% of the 14q32 translocations, various other partners are identified only rarely, so that the translocation usually is designated 14q+ (3, 4, 5). Because MM cells have undergone IgH isotype switching, we postulated that as in mouse plasmacytomas (6), the translocations would occur into IgH switch regions.
The order of the constant region genes in the IgH locus, starting from the telomeric end, is μ, δ, γ3, γ1, ψɛ, α1, ψγ, γ2, γ4, ɛ, and α2. Isotype switch from μ to γ, α, or ɛ is a developmentally regulated DNA recombination event that occurs between Sμ and the respective isotype switch region, resulting in the formation of a hybrid switch region. The switch regions are 1–3 kb long, consist of tandem pentameric repeats, and are located just upstream of μ, γ3, γ1, α1, γ2, γ4, ɛ, and α2 constant region segments. There is no conventional switch region upstream of δ, ψɛ, or ψγ (7). However, isotype recombination to δ can occur by homologous recombination between duplicated 442-bp sequences (σμ and σδ) that are located upstream of Sμ and the first δ exon, respectively (8, 9).
A “legitimate” IgH switch recombination occurs between two IgH switch regions, resulting in the formation of a hybrid switch region. This may be a productive switch from μ to another isotype, a downstream switch not involving μ, or an inversion event between switch regions. In most cases, these recombination events occur on the same chromosome (cis), but occasionally occur in trans (recombination between sister chromatids or homologous chromosomes). “Illegitimate” IgH switch recombination, which is defined as involving only one IgH switch region, may be a chromosomal translocation, an interstitial deletion, an interstitial insertion, or an inversion into a switch region. We have developed a comprehensive Southern blot assay that can distinguish the different kinds of legitimate and illegitimate switch recombination. Using this assay, we have screened a panel of MM cell lines and primary tumor samples to determine the kinds of switch recombination events that occur in this neoplasm. We have found that a high fraction contains illegitimate switch recombination fragments, many of which identify chromosomal translocations.
MATERIALS AND METHODS
Cell Culture.
The human MM cell lines were grown in Petri dishes in RPMI 1640 medium supplemented with 10% fetal calf serum. The 8226 (10) and U266 (11) lines were obtained from American Type Culture Collection. Lines H929 (12) and H1112 were established at our institution (National Cancer Institute) by H. Oie. The remaining cell lines were generously provided to us by many investigators from around the world. SK-MM1 and SK-MM2 (13) were from A. Houghton (New York); KMM-1, KMS-11, and KMS-12 (14) were from M. Namba (Okayama, Japan); OPM-2 (15) was from S. Katagiri (Osaka) care of B. Thompson (Galveston, TX); MM-M1 (16) was from T. Takahashi (Kobe, Japan); JJN3 (17) and JIM3 (18) were from I. Franklin (Glasgow, U.K.); ark (19) was from J. Epstein (Little Rock, AR); ANBL-6 (20) was from D. Jelinek (Rochester, MN); OCI-My-5 (21) was from D. Bergsagel (Toronto); UTMC-2 (22) was from A. Solomon (Knoxville, TN); Delta-47 (23, 24) and FLAM-76 were from I. Kubonishi (Kochi, Japan); MM.1 (25) was from N. Krett (Chicago); and TH (26) was from R. van Lier (Amsterdam).
Karyotypes.
Chromosomal analysis was performed on the ark and MM-M1 cell lines by a G-banding method (Molecular Medicine, Rockville, MD). The SK-MM2 karyotype is an unpublished result of R. S. K. Chaganti (personal communication), and the other karyotypes are referenced above.
Mapping of Translocation Breakpoints.
PCR primers were designed to amplify the sequence from the unknown end (i.e., the end that did not contain sequences from the selected switch region) of the illegitimate switch region fragment clone. PCR analysis was performed on the following mapping panels: (i) NIGMS Mapping Panel 2 (Coriell Cell Repository, Camden, NJ), each somatic cell hybrid contains a single human chromosome; (ii) BIOS mapping panel, each hybrid contains several human chromosomes; (iii) GENEBRIDGE4 radiation hybrid panel (Research Genetics, Huntsville, AL) (27); and (iv) an 11q13-specific panel of radiation hybrids (28, 29). Sublocalization was done by fluorescence in situ hybridization, sequence identity to known chromosomal regions, or mapping to cosmid or yeast artificial chromosome clones containing genomic sequences (30, 31, 32).
Other Procedures.
Southern blots, PCR analysis, probes, hybridization, preparation and analysis of genomic libraries, and DNA sequencing are described elsewhere (31).
RESULTS
A Southern Blot Assay to Distinguish Legitimate and Illegitimate Switch Recombination Fragments.
We designed five pairs of probes (Table 1) that hybridize upstream and downstream of the sequences known to be important in mediating IgH switch recombination: σμ/σδ, Sμ, Sγ, Sα, and Sɛ. Southern blots are generated using restriction endonucleases that digest DNA at sites outside the pair of switch probes. Different recombination events are reflected in different patterns of cohybridization of the flanking probes as diagrammed in Fig. 1. As shown, this assay can distinguish among different kinds of legitimate switch recombination events, including different results for cis versus trans recombination (the latter illustrated for productive switch recombination by the retention of a second legitimately rearranged fragment that is lost during cis recombination). Importantly, a candidate illegitimate switch recombination fragment is easily recognized as a restriction fragment to which only 1 of the 10 switch probes hybridizes.§
Table 1.
Sequence of PCR primers used to amplify switch region probes
| Probe | Forward primer | Reverse primer | Size, bp | Annealing temperature, °C |
|---|---|---|---|---|
| 5′σμ | CAGATCTGAAAGTGCTCTACTG | CTTATCCTAAAGTGAGTAGTTG | 462 | 50 |
| 3′σδ | CACCGAAACCTCTGGAGGGAAG | TGTGCTGGACCACGCATTTG | 548 | 63 |
| 5′Sμ | TTTGAAGGAGAGGTCGCACGAG | TCAGCTAAAGCCATCTCATTGCC | 631 | 58 |
| 3′Sμ | TCTACACTGCGTTCCCCATCAC | CGTTCTGAGTGCCCTCACTACTTG | 593 | 58 |
| 5′Sγ | CAGAATGGTCATAATCGCTGCC | CATCCCGTCATGTTCCTCGTG | 719 | 58 |
| 3′Sγ | GCTATTCCAAGACAGGGGGTTCC | CAGAAAGCTTGCAGGACCG | 436 | 55 |
| 5′Sα | CAGCATCCAGCCACATCTG | AGATCCTTCCTGCCTGGTTAG | 405 | 58 |
| 3′Sα | CATGGTGCAGGAGCTGTGTAAC | TCCACTCTGGTGTGAGTGAAGG | 562 | 58 |
| 5′Sɛ | AAGAGAACCTCCCCAGCACTC | GTCGGGTCTCTGACTTCTTGGTCT | 558 | 61 |
| 3′Sɛ | CGGGGCTGGATACTGTGATTTTG | TATCATCAGGCTGGGCTCAGGAAG | 547 | 58 |
The sequence of the primers is given from 5′ to 3′.
Figure 1.
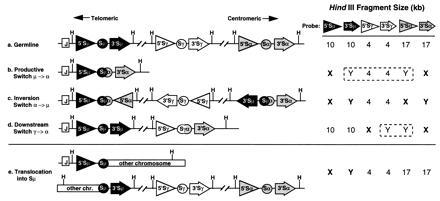
Identification of IgH switch recombination events. A schematic map showing the organization of three IgH switch region loci is shown. The μ (solid), γ (open), and α (shaded) switch region loci are illustrated, with critical elements depicted as follows: circle, switch region; triangle, 5′ switch probe; arrow, 3′ switch probe; and vertical line (H), HindIII restriction enzyme sites. Structural configurations in the germ line and after various recombination events are indicated. The relevant sizes of typical germ-line HindIII restriction fragments detected by each switch probe are shown. The fragments enclosed within dashed lines are deleted during cis recombination but retained during trans recombination. After recombination, germ-line fragments are replaced by rearranged fragments (X and Y) that have different patterns of hybridization with the 5′ and 3′ switch probes, depending on the kind of recombination event. The direction of the triangle and arrow indicates the orientation of transcription, which is unchanged except in the case of inversion.
The location of the probes, the relevant restriction enzyme sites, and Southern blots of germ-line DNA hybridized to these probes are shown in Fig. 2. As expected, each pair of flanking probes hybridize to the same respective restriction fragment(s). Only a single restriction fragment is identified respectively for the σ, μ, and ɛ probe pairs (the ψɛ sequence is not detected by the ɛ probe pair). The Sγ probes cohybridize to γ1, γ2, γ3, and γ4 but not to ψγ. The five Sγ fragments evident in this sample are presumably due to a polymorphism that distinguishes the length of a particular maternal versus paternal Sγ. The Sα probes cohybridize to both α1 and α2. The 3′Sα probe is bisected by a SphI site in α2, so that it hybridizes to 3.7-kb and 2.0-kb α2 SphI fragments, with only the 3.7-kb fragment cohybridizing with 5′Sα.
Figure 2.
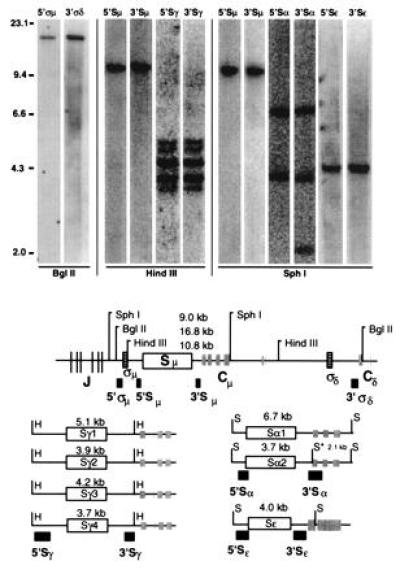
(Upper) Southern blot analysis of germ-line IgH switch regions. Placental genomic DNA was digested with the indicated restriction enzyme, subjected to electrophoresis on a 0.7% agarose gel, blotted, probed sequentially with various switch probes, and scanned on a PhosphorImager (Molecular Dynamics). The probe is shown above each lane, and lambda HindIII markers are indicated at the left. (Lower) The positions of switch regions (open rectangles), sigma sequences (boxes with horizontal lines), joining region and constant region exons (shaded boxes and lines), 5′ and 3′ switch probes (filled boxes), and restriction sites (S, SphI; B, BglII; H, HindIII) are indicated. The asterisk indicates a SphI site that is present in the 3′ Sα probe and the alpha-2 locus, but not the alpha-1 locus. With the exception of one fragment detected uniquely by the 3′ Sα probe (see text), all 5 pairs of probes cohybridize to the same respective fragment(s).
Illegitimate IgH Switch Recombination Fragments Are Detected in Most MM Cell Lines.
Table 2 summarizes our results for 21 MM cell lines, 12 of which have karyotypic translocations involving 14q32. All but one line (FLAM-76) has at least one legitimate or illegitimate switch recombination fragment. Most of the legitimate switch recombination fragments are classified as productive (Introduction and Fig. 1) and are fully consistent with the isotype of IgH expressed by the cell line. In three cases (KMS-12, ark, and Delta-47), there is evidence of a downstream switch recombination fragment that includes Sγ and Sα sequences. In one case (ark), there is evidence of an inversion between a productive Sμγ and Sα.
Table 2.
IgH switch recombination: Summary of 21 human MM cell lines
| Line | Isotype*
|
14q32 abnormality† | Legitimate switch‡ | Illegitimate switch§¶
|
||
|---|---|---|---|---|---|---|
| H | L | 5′ probe | 3′ probe | |||
| FLAM-76 | — | κ | 11q13‖ | None | None | None |
| MM-M1 | (γ) | (κ) | 11q13‖ | Sμ→σδ, Sμ→Sγ | None | None |
| H1112 | δ | λ | 11‖ | Sμ→σδ, Sμ→Sγ | None | None |
| SK-MM2 | — | κ | 11q13‖ | None | None | Sγ [11q13] |
| KMS-12 | NP | 11q13‖ | Sμ→σδ, Sα→Sγ | Sμ, Sα | Sγ [11q13], Sμ [14]** | |
| Delta-47 | δ | λ | 14q+ | Sμ→σδ, Sμ→Sα, Sγ→Sα | None | None |
| TH | ㇇ | κ | 14q+ | Sμ→Sα | Sμ | None |
| 8226 | (γ) | λ | 1p13 | None | None | Sㆆ |
| MM.1 | (α) | λ | 12q24 | None | Sμ | Sγ |
| JJN3§§ | (α) | κ | 14q+ | None | 5′σμ | Sμ [16q23]§§ |
| SK-MM1 | — | κ | 14q+ | Sμ→Sγ | Sμ | Sμ [6], Sγ |
| JIM3§§ | (α) | λ | 14q+ | None | Sμ | Sγ [4p16]§§ |
| KMS-11 | (γ) | κ | None | None | Sμ | Sμ [4p16], Sμ, Sγ |
| KMM-1 | — | λ | None | None | σμ [8q24] | σδ [21q22] |
| U266 | ɛ | λ | None‖ | Sμ→Sɛ | Sγ | Sα [11q13] |
| H929 | (α) | κ | None | None | Sμ | Sα |
| OPM-2 | (γ) | λ | None | None | Sμ | Sγ |
| ANBL-6 | — | λ | None | None | Sμ | Sμ |
| UTMC-2 | α | κ | None | Sμ→Sα | Sμ | Sμ |
| ark | γ | κ | None | Sμ→Sγ, Sα→Sγ, Sα↔Sμγ | None | None |
| OCI-MY5 | α | λ | ND | Sμ→Sα | None | None |
Isotype of IgH chain present in tumor but absent from cell line indicated by parentheses, from original reference or investigator, as cited. NP, Nonproducer.
Karyotype from original reference as cited, except ark, SK-MM2, and MM-M1; H1112 has an 11;14 translocation by chromosome painting analysis (M.C. and E. Schrock, unpublished data); ND, Not done.
A legitimate switch recombination fragment hybridizes to two switch region probes (see Fig. 1). Productive and downstream switch indicated by →; inversion indicated by ↔.
Cyclin D1 mRNA overexpressed in these lines.
An illegitimate switch recombination fragment hybridizes only to a single 5′ (column 5) or 3′ (column 6) probe and to no other probes.
Chromosomal partner in cloned breakpoint indicated within square brackets.
Unidentifed sequence from chromosome 14 juxtaposed to Sμ.
This cell line expresses alpha mRNA, not gamma mRNA (data not shown).
This illegitimate switch was not detected by any of the 10 probes in Table 1.
By Southern blot analysis the translocation breakpoint identified by this probe was identified in primary tumor corresponding to this cell line.
In 15 of 21 lines, we have identified one or more apparent illegitimate switch recombination fragments that are candidates for switch-mediated translocation breakpoints, including seven of eight lines in which no karyotypic abnormality of 14q32 has been identified. Strikingly, in three of the six lines lacking a detectable illegitimate switch fragment, there is a karyotypic t(11;14) translocation and overexpression of cyclin D1 (see Discussion). As might be expected with a reciprocal translocation, a single illegitimate switch recombination event generates two illegitimate switch recombination fragments (Fig. 1e), although both may not be detected because of loss from the cell and/or the technical limitations of Southern blotting. For most MM cell lines, one (three lines) or two (nine lines) illegitimate switch recombination fragments are detected. In general, when two illegitimate switch fragments are detected, one fragment is identified by a 5′Sμ probe and the other identified by a 3′ Sμ, 3′Sγ, or 3′Sα probe. Thus, there is evidence for a single illegitimate switch recombination event in 12 MM lines. However, three lines have more than two illegitimate switch recombination fragments (three in SK-MM1, four in KMS-11, and four in KMS-12), suggesting that two illegitimate switch recombination events may have occurred. Examples of MM lines without any (MM-M1) and with two (KMM-1) illegitimate switch recombination fragments are shown in Fig. 3 and described below.
Figure 3.
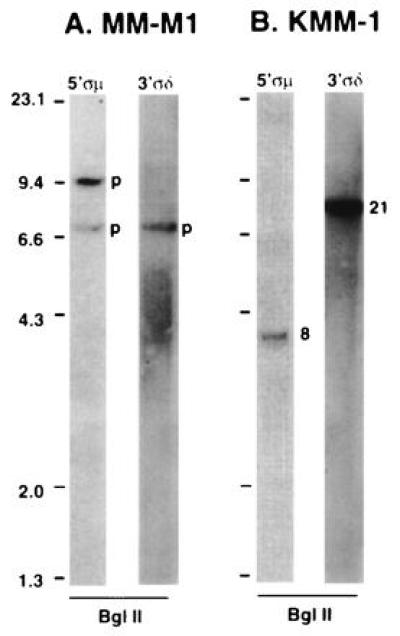
Southern blot analysis of switch regions in two MM cell lines. Genomic DNA from two cell lines was digested with the indicated restriction enzyme, and Southern blots were analyzed sequentially with various switch probes as described in Fig. 2. The probe is indicated above each lane. (A) MM-M1 cell line. (B) KMM-1 cell line. Illegitimate switch recombination fragments are indicated by 8 and 21 (corresponding to the chromosome involved in the recombination). Productive recombination fragments are indicated by a p.
Legitimate and Illegitimate Recombinations into σμ/σδ Sequences.
As stated above, isotype recombination from μ to δ heavy chain can occur by homologous recombination between σμ and σδ sequences that are located upstream of Sμ and IgH δ, respectively (Fig. 2). Examples of Southern blots for two MM cell lines, in each case using BglII-digested genomic DNA that was probed sequentially with 5′σμ and 3′σδ probes, are shown in Fig. 3.
For the MM-M1 cell line (Fig. 3A), the 5′σμ probe hybridizes to 7-kb and 9.6-kb BglII fragments, whereas the 3′σδ probe hybridizes only to the 7-kb fragment. The 9.6-kb fragment is derived from a productive μ to γ legitimate switch recombination event (data not shown for γ). The 7-kb BglII fragment that cohybridizes with the two probes is smaller than the germ-line fragment (16.8 kb; Fig. 2) and larger than the ≈2-kb fragment generated by recombination between σμ and σδ to mediate a stable μ to δ switch (9). Further mapping with a number of restriction enzymes indicates that MM-M1 has a recombination event that deletes about 10 kb of sequences starting within Sμ and ending about 2 kb upstream of σδ. Thus, MM-M1 appears to have an Sμ/σδ recombination event that is predicted to be functionally similar to the stable μ to δ productive isotype switch mediated by recombination between the homologous σμ and σδ sequences. Although MM-M1 has a t(11;14)(q13;q32) translocation, we were unable to identify an illegitimate switch recombination fragment that identifies a potential translocation event (see below).
For the KMM-1 cell line, the 5′σμ probe hybridizes to a 3.6-kb fragment (indicated by an 8 in Fig. 3B) and the 3′σδ probe hybridizes to a 7.5 kb-fragment (indicated by 21 in Fig. 3B), indicating dissociation of these two probes that cohybridize to a 16.8-kb germ-line fragment in placental DNA (Fig. 2). We have cloned both fragments and confirmed by sequence analysis that each represents a translocation breakpoint. The 5′ end of the 3.6-kb BglII fragment contains germ-line sequences 5′ of σμ but is disrupted within the σμ sequence, and contains unknown sequences from chromosome 8 at its 3′ end. The 7.5-kb fragment contains the expected δ and σδ sequences at its 3′ end but is disrupted about 400 bp upstream of σδ, and contains unknown sequences from chromosome 21 at its 5′ end. The KMM-1 MM cell line thus represents an example of a complex chromosomal translocation into or near sequences (i.e., σμ and σδ) that are known to be capable of mediating, by homologous recombination, a switch from μ to δ.
Translocation Breakpoints Have Been Isolated for All Eight MM Lines from Which Illegitimate Switch Recombination Fragments Have Been Cloned.
We have isolated molecular clones containing illegitimate switch recombination fragments from eight lines and have confirmed the presence of a translocation into a switch region in each line by mapping the chromosomal origin of the non-Ig sequence (Tables 2 and 3). In all eight lines, we isolated translocation breakpoint clones with the novel chromosomal partner telomeric to the 3′ IgH sequences on chromosome 14 (cf. Fig. 1, bottom line of e). In one line (KMM-1), we also isolated a translocation breakpoint clone with the 5′ IgH sequences telomeric to the novel chromosomal partner (8q24) (cf. Fig. 1, top line of e). For three lines that overexpress cyclin D1 (U266, KMS-12, and SK-MM2) (31), a switch region is juxtaposed to chromosome 11q13 sequences. For five other lines, the switch regions are juxtaposed to five different chromosomes: 4 in JIM3 and KMS-11, 6 in SK-MM1, 8 and 21 in KMM-1, and 16 in JJN3. Except for the lines with cyclin D1 overexpression, we do not know what genes are dysregulated by the translocations.
Illegitimate Switch Recombination Fragments Are Detected in Primary Tumor Samples.
Examples of Southern blots that illustrate apparent illegitimate switch recombination events in tumor cells from two patients are shown in Fig. 4. Informative results using a nearly homogeneous population of IgG plasma cell leukemia tumor cells (patient 1) are shown in Fig. 4A. The karyotype of these cells was markedly abnormal, although no abnormalities of 14q32 were noted. A 5′Sμ probe hybridizes equally to 2.5-kb (indicated by a 4 in Fig. 4A) and 3.0-kb (indicated by a p in Fig. 4A) HindIII fragments but identifies only traces of a germ line 10.6-kb fragment contributed by normal cells. The 3.0-kb fragment, which cohybridizes with a 3′Sγ probe, probably represents a productive legitimate switch recombination. In contrast, the 2.5-kb fragment that hybridizes to a 5′Sμ probe and the 5.4-kb fragment (indicated by ∗ in Fig. 4A) that hybridizes to a 3′Sγ probe represent illegitimate switch recombination fragments because they are not identified by other switch probes. The illegitimate switch fragment identified by the 5′Sμ probe (4 in Fig. 4A) was cloned and found to represent a translocation breakpoint with chromosome 4 sequences juxtaposed to Sμ sequences.
Figure 4.
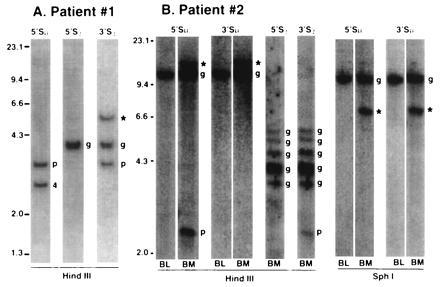
Southern blot analysis of switch regions in two patients with MM. Genomic DNA from two tumor samples was digested with the indicated restriction enzyme, and Southern blots were analyzed sequentially with various switch probes as described in Fig. 2. The probe is indicated above each lane. (A) Patient 1. (B) Patient 2. For patient 2, DNA was prepared from bone marrow (BM) or blood (BL). Illegitimate switch recombination fragments are indicated by 4 (corresponding to the chromosome involved in the recombination), and ∗ if it has not been cloned. Productive recombination fragments are indicated by a p, and germ-line fragments by a g.
A bone marrow sample that contains about 30% tumor cells was obtained from a second patient with intramedullary IgG MM (Fig. 4B). Peripheral blood mononuclear cells from that patient served as a control. A 2.3-kb fragment (indicated by a p in Fig. 4B) that cohybridizes uniquely with 5′Sμ and 3′Sγ probes is present only in the bone marrow and likely represents a productive legitimate switch recombination. Both 5′Sμ and 3′Sμ probes identify an intense germ line 10.6-kb HindIII (9.6-kb SphI) fragment (indicated by a g in Fig. 4B) in bone marrow and blood. However, a substantially less intense 13.4-kb HindIII (6.4-kb SphI) fragment (indicated by ∗ in Fig. 4B) is detected only in bone marrow DNA and must represent an illegitimate switch recombination event in the tumor (despite the fact that the 5′ and 3′ Sμ probes are not dissociated). The cohybridization of the 5′ and 3′ Sμ probes to a larger HindIII fragment (indicated by ∗ in Fig. 4B) and a smaller SphI fragment (compared with germ line) indicates that the DNA rearrangement did not occur within Sμ but occurs near Sμ, i.e., either within a HindIII site about 1 kb upstream or within a SphI site about 2.5 kb downstream of Sμ.
DISCUSSION
Previous cytogenetic studies have detected 14q32 translocations in 10–60% of MM patients (3, 4, 5). Yet with the exception of involvement of 11q13 in about 30% of the 14q32 translocations in MM, a variety of other partners have been identified rarely, so the translocations are typically designated 14q+. Because human MM cells are thought to be a malignant version of long-lived bone marrow plasma cells that have been productively selected by antigen in germinal centers and have undergone extensive somatic hypermutation of Ig genes and IgH switching (1, 33, 34, 35), we postulated that as in mouse plasmacytomas (6), the translocations would occur into IgH switch regions. Therefore, we designed a Southern blot assay that enabled us to identify apparent illegitimate switch recombination fragments (candidate translocation breakpoints) in 15 of 21 MM cell lines. From each of the eight lines and one patient sample that we analyzed further, we have identified a translocation breakpoint by cloning an illegitimate switch recombination fragment. Moreover, we found (Table 3 and data not shown) that 10 of 12 illegitimate switch fragments that were cloned are, in fact, IgH switch translocation breakpoints involving another chromosome (the exceptions included a polymorphism and a rearrangement to an unknown portion of chromosome 14). Importantly, illegitimate switch recombination fragments have been identified in seven of eight MM lines that do not have a karyotypically detectable 14q32 translocation. Thus, we have developed a more sensitive technology that has enabled us to identify apparent translocations into IgH switch regions in a majority of MM cell lines and tumors.
Table 3.
Mapping of translocation breakpoints
| Line | Locus | Method | Primers |
|---|---|---|---|
| SK-MM2 | 11q13.3 | YAC yWPR11 | GGATGGTTCTGTGTTATAGTCTG |
| GCAATGAAGCCAGGTCAG | |||
| KMS-12 | 11q13 | 11q13 RH | GACCTTGCAGTGACAAAACCAAG |
| CCCCACTTTTAGGAACCATAGCTTC | |||
| U266 | 11q13.3 | YAC y14F8 | CGCAAACATGGCAGTGACTTTTC |
| CCTGTCTTCAAGGAAACCACTCCC | |||
| JJN3 | 16q23.1 | YAC y798A3 | TGGTAACATCAGGCCAAGTCTCT |
| TTACATGGCAGATGGGGAATTAAGG | |||
| SK-MM1 | 6 | NIGMS SCH | ACACAGAGGAGTGTCTGTGAAGA |
| GGAGTGTTAGTTACCCTATATCCC | |||
| KMM-1 | 8q24.13 | GB4 RH | GATCTTGGCTGAGATCGGTAGGA |
| CCGTGCAAAATTAAATCCTCAC | |||
| KMM-1 | 21q22.1 | GB4 RH | AGCTTGCCCCAAACACTTCT |
| CTCTGCAGAAAGGCCATTCTCAAA | |||
| JIM3 | 4p16.3 | Cosmid 184d6 | AAGCTTGTTTCTTAACGTGGTTCT |
| KMS-11 | 4p16.3 | Cosmid 75b9 | AAGCTTGGTGGAACGTGTCCACG |
| Patient 1 | 4p16.3 | Cosmid 75b9 | AAGCTTACACAAACTCAAAATTG |
Using PCR primers, the non-Ig sequence was mapped to a chromosome using somatic cell hybrid (SCH) panels. Mapping to a chromosomal locus is based on results with radiation reduced hybrid (RH) panels or clones containing genomic sequences. The JJN3 breakpoint was also mapped by fluorescein in situ hybridization analysis. For JIM3, KMS-11, and patient 1, the non-Ig sequence was mapped to cosmids 184d6 and 75b9 by sequence identity with the indicated primer site.
Because four of five lines for which we did not detect any illegitimate switch recombinations have a karyotypically identified 14q32 translocation (a sixth line, OCI-MY5, has not been karyotyped), it is possible that, as in other B lymphoma tumors, some translocations in MM are mediated by aberrant VDJ recombination and occur into joining region genes. Alternatively, it is possible that our assay, which uses five pairs of probes, is unable to detect every translocation into or near switch regions. For cell line 8226, for example, we detected no illegitimate switch recombination fragments, but because we noted that this cell line had deleted joining region genes, μ, and δ, by further mapping we were able to localize an illegitimate recombination into a γ region but not near a switch site (Table 2).
The IgH translocations in MM cell lines clearly are not solely associated with cell culture, because (i) translocations involving the 14q32 locus have been identified in 10–60% (in different studies) of MM tumors; (ii) we have identified illegitimate switch recombination fragments in MM tumors (Fig. 4), and the one we have cloned is, in fact, a translocation breakpoint; and (iii) for the two lines for which we have analyzed the corresponding primary tumor, the translocation breakpoint is also present in the primary tumor (Table 2).
Are the IgH switch translocations in MM an early event in pathogenesis or do they occur during tumor progression? Although we do not have a definitive answer to this question, there is some information regarding the question of whether switch recombination occurs in MM cells. First, when MGUS progresses to MM, which occurs at a rate of about 1% per year, the IgH isotype is unchanged (R. Kyle, personal communication); this suggests that a sequential productive switch recombination does not occur at a high rate in premalignant MGUS. Second, the IgH isotype almost always remains the same through the various stages of MM tumor progression, although there are rare cases in which a clonal isotype switch has been reported (36). Third, in mouse plasmacytoma lines that have a phenotype that is similar to, but not identical to, human MM tumors, sequential productive IgH switching is rare, occurring at rates of 10−5 per cell per generation at most (37, 38). Despite our inability to provide a definitive answer to this question at present, we think it is likely that IgH switch translocations are an early event in the pathogenesis of MM, occurring concordantly with physiological switch recombination in a germinal center B cell and perhaps providing the initial event in generating premalignant MGUS.
Chromosomal translocations to the IgH locus provide a seminal molecular pathogenetic event in many B cell malignancies. Often, there is a predominant chromosomal locus (and oncogene) that is translocated to the IgH locus for each type of B cell tumor [e.g., bcl-1 in mantle cell lymphoma (39), bcl-2 in follicular lymphoma (40), and c-myc in Burkitt’s lymphoma (41) or mouse plasmacytoma (42)]. Less frequently, there is a predominant oncogene that is dysregulated by translocation to a variety of other chromosomal loci, which can include the IgH locus [bcl-6 at 3q27 in diffuse lymphoma (43) and MLL at 11q23 in acute leukemias (44)]. Strikingly, the 10 switch translocation breakpoints we have cloned from eight MM lines and one MM tumor involve six different loci: 4p16 (two lines and the one tumor); 6; 8q24; 11q13 (in three lines), 16q23, and 21q22. The location of the cloned breakpoints at telomeric sites (4p16, 16q23, and 21q22) is consistent with the fact that in MM translocations are often designated 14q+ because a partner chromosome cannot be specifically identified. In any case, translocations into the IgH locus in MM are not only frequent but also involve two recurrent translocation partners (11q13 and 4p16) as well as an apparently highly diverse array of translocation partners: 1p13, 6, 8q24, 12q24, 16q23, and 21q22 (Table 2) in the cell lines we analyzed, and 1q21, 3p11, 6p21, 7q11, 8q24 (c-myc), 11q23 (MLL), and 18q21 (bcl-2) from published karyotypes of tumors (4, 5). The promiscuous array of chromosomal translocation partners that are thought to activate bcl-6 or MLL (see above) stands in contrast to the promiscuous array of chromosomal translocation partners juxtaposed to the IgH locus in MM. The basis for this apparent diversity of translocation partners is unclear. It is possible that some of the translocations involving nonrecurrent, diverse loci are a consequence of genetic instability affecting IgH switch regions in MM and do not contribute directly to the malignant phenotype. However, we favor the hypothesis that a cell that is already committed to differentiation into a long-lived plasma cell is particularly susceptible to malignant transformation by any of a number of different dysregulated or altered oncogenes, all resulting in a similar phenotype.
Based on the results of large studies of karyotypes in MM and on the results presented here, there are several obvious implications. First, cytogenetics grossly underestimates the frequency of 14q32 translocations in MM, which appear to be a nearly universal event in the MM cell lines we examined. Second, based on the type of translocation, it may be possible, as in lymphoma, to divide MM into subtypes that may have different clinical courses or therapeutic sensitivities. In fact, there is recently published evidence that MM tumors with t(11;14)(q13;q32) have a distinct morphology (small plasma cells, often cleaved) (45), and that MM tumors with 11q abnormalities carry a poor prognosis (46). Third, clonally related premalignant MGUS and MM, which share the same productive IgH switch event, may also share a common translocation event into an IgH switch region. Finally, 14q32 translocations are the most consistent genetic abnormality to be described in MM. As in lymphoma, we hypothesize that these translocations activate a variety of oncogenes. Identification of these genes promises to greatly enhance our understanding of the pathogenesis of MM.
Acknowledgments
We thank Fred C. Mills (National Cancer Institute) for sharing sequences of γ switch regions before publication, Evelin Schrock (National Center for Human Genome Research) for assisting us with fluorescent in situ hybridization analysis, Norman Doggett for assisting with 16q23 mapping, and Ilan Kirsch for helpful discussions about translocations. We are grateful for the MM cell lines kindly provided to us by many investigators.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: MM, multiple myeloma; MGUS, monoclonal gammopathy of undetermined significance; IgH, immunoglobulin heavy chain; Sμ, switch μ.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base (accession nos. U73660–U73677, U73660, U73661, U73662, U73663, U73664, U73665, U73666, U73667, U73668, U73669, U73670, U73671, U73672, U73673, U73674, U73675, U73676, U73677).
Fragments that hybridize with only one switch probe could be generated without switch recombination, by deletion of sequences corresponding to one of a pair of switch probes, although we have not encountered this among 12 examples that have been cloned and sequenced.
References
- 1.Malpas J S, Bergsagel D E, Kyle R. Multiple Myeloma: Biology and Management. Oxford: Oxford Univ. Press; 1995. [Google Scholar]
- 2.Kyle R A. Mayo Clin Proc. 1993;68:26–36. doi: 10.1016/s0025-6196(12)60015-9. [DOI] [PubMed] [Google Scholar]
- 3.Lai J L, Zandecki M, Mary J Y, Bernardi F, Izydorczyk V, Flactif M, Morel P, Jouet J P, Bauters F, Facon T. Blood. 1995;85:2490–2497. [PubMed] [Google Scholar]
- 4.Sawyer J R, Waldron J A, Jagannath S, Barlogie B. Cancer Genet Cytogenet. 1995;82:41–49. doi: 10.1016/0165-4608(94)00284-i. [DOI] [PubMed] [Google Scholar]
- 5.Taniwaki M, Nishida K, Takashima T, Nakagawa H, Fujii H, Tamaki T, Shimazaki C, Horiike S, Misawa S, Abe T, Kashima K. Blood. 1994;84:2283–2290. [PubMed] [Google Scholar]
- 6.Potter M, Wiener F. Carcinogenesis. 1992;13:1681–1697. doi: 10.1093/carcin/13.10.1681. [DOI] [PubMed] [Google Scholar]
- 7.Harriman W, Volk H, Defranoux N, Wabl M. Annu Rev Immunol. 1993;11:361–384. doi: 10.1146/annurev.iy.11.040193.002045. [DOI] [PubMed] [Google Scholar]
- 8.White M B, Word C J, Humphries C G, Blattner F R, Tucker P W. Mol Cell Biol. 1990;10:3690–3699. doi: 10.1128/mcb.10.7.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yasui H, Akahori Y, Hirano M, Yamada K, Kurosawa Y. Eur J Immunol. 1989;19:1399–1403. doi: 10.1002/eji.1830190808. [DOI] [PubMed] [Google Scholar]
- 10.Matsuoka Y, Moore G E, Yagi Y, Pressman D. Proc Soc Exp Biol Med. 1967;125:1246–1250. doi: 10.3181/00379727-125-32327. [DOI] [PubMed] [Google Scholar]
- 11.Nilsson K, Bennich H, Hohansson S G, Ponten J. Clin Exp Immunol. 1970;7:477–489. [PMC free article] [PubMed] [Google Scholar]
- 12.Gazdar A F, Oie H K, Kirsch I R, Hollis G F. Blood. 1986;67:1542–1549. [PubMed] [Google Scholar]
- 13.Eton O, Scheinberg D A, Houghton A N. Leukemia. 1989;3:729–735. [PubMed] [Google Scholar]
- 14.Namba M K, Ohtsuki T, Mori M, Togawa A, Wada H, Sugihara T, Yawata Y, Kimoto T. In Vitro Cell Dev Biol. 1989;25:723–729. doi: 10.1007/BF02623725. [DOI] [PubMed] [Google Scholar]
- 15.Katagiri S, Yonezawa T, Kuyama J, Kanayama Y, Nishida K, Abe T, Tamaki T, Ohnishi M, Tarui S. Int J Cancer. 1985;36:241–246. doi: 10.1002/ijc.2910360217. [DOI] [PubMed] [Google Scholar]
- 16.Okuno Y, Takahashi T, Suzuki A, Ichiba S, Nakamura K, Fukumoto M K, Okada T, Okada H, Imura H. Leukemia. 1991;5:585–591. [PubMed] [Google Scholar]
- 17.Jackson N, Lowe J, Ball J, Bromidge E, Ling N R, Larkins S, Griffith M J, Franklin I M. Clin Exp Immunol. 1990;75:93–99. [PMC free article] [PubMed] [Google Scholar]
- 18.Barker H, Hamilton M, Ball J, Drew M, Franklin I. In: EURAGE Monoclonal Gammopathies III: Clinical Significance and Basic Mechanisms. Radl J, van Camp B, editors. Leiden, The Netherlands: EURAGE; 1991. pp. 155–158. [Google Scholar]
- 19.Ridley R C, Xiao H, Hata H, Woodliff J, Epstein J, Sanderson R D. Blood. 1993;81:767–774. [PubMed] [Google Scholar]
- 20.Jelinek D F, Ahmann G J, Greipp P R, Jalal S M, Westendorf J J, Katzmann J A, Kyle R A, Lust J A. Cancer Res. 1993;53:5320–5327. [PubMed] [Google Scholar]
- 21.Hitzler J K, Martinez-Valdez H, Bergsagel D E, Minden M D, Messner H A. Blood. 1991;78:1996–2004. [PubMed] [Google Scholar]
- 22.Ozaki S, Wolfenbarger D, de Bram-Hart M, Kanangat S, Weiss D T, Solomon A. Leukemia. 1994;8:2207–2213. [PubMed] [Google Scholar]
- 23.Ishii K, Yamato K, Kubonishi I, Taguchi H, Ohtsuki Y, Miyoshi I. Am J Hematol. 1992;41:218–224. doi: 10.1002/ajh.2830410314. [DOI] [PubMed] [Google Scholar]
- 24.Kubonishi I, Seto M, Shimamura T, Enzan H, Miyoshi I. Cancer. 1992;70:1528–1535. doi: 10.1002/1097-0142(19920915)70:6<1528::aid-cncr2820700614>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 25.Goldman-Leikin R E, Salwen H R, Herst C V, Variakojis D, Bian M L, Le Beau M M, Selvanayagan P, Marder R, Anderson R, Weitzman S, Rosen S T. J Lab Clin Med. 1989;113:335–345. [PubMed] [Google Scholar]
- 26.Weinreich S S, von dem Borne A E, van Lier R A, Feltkamp C A, Slater R M, Wester M R, Zeijlemaker W P. Br J Haematol. 1991;79:226–234. doi: 10.1111/j.1365-2141.1991.tb04526.x. [DOI] [PubMed] [Google Scholar]
- 27.Walter M A, Spillett D J, Thomas P, Weissenbach J, Goodfellow P N. Nat Genet. 1994;7:22–28. doi: 10.1038/ng0594-22. [DOI] [PubMed] [Google Scholar]
- 28.Smith C M, Bora P S, Bora N S, Jones C, Gerhard D S. Cytogenet Cell Genet. 1995;71:235–239. doi: 10.1159/000134117. [DOI] [PubMed] [Google Scholar]
- 29.Gerhard D S, Lawrence E, Wu J, Chua H, Ma N, Bland S, Jones C. Genomics. 1992;13:1133–1142. doi: 10.1016/0888-7543(92)90028-q. [DOI] [PubMed] [Google Scholar]
- 30.Baxendale S, MacDonald M E, Mott R, Francis F, Lin C, Kirby S F, James M, Zehetner G, Hummerich H, Valdes J, Collins F S, Deaven L J, Gusella J F, Lehrach H, Bates G P. Nat Genet. 1993;4:181–186. doi: 10.1038/ng0693-181. [DOI] [PubMed] [Google Scholar]
- 31.Chesi M, Bergsagel P L, Brents L A, Smith C A, Gerhard D S, Kuehl W M. Blood. 1996;88:674–681. [PubMed] [Google Scholar]
- 32.Doggett N A, Goodwin L A, Tesmer J G, Meincke L J, Bruce D C, Clark L M, Altherr M R, Ford A A, Chi H C, Marrone B L. Nature (London) 1995;377:335–365. doi: 10.1038/377335a0. [DOI] [PubMed] [Google Scholar]
- 33.Bakkus M H, Heirman C, Van Riet I, Van Camp B, Thielemans K. Blood. 1992;80:2326–2335. [PubMed] [Google Scholar]
- 34.Vescio R A, Cao J, Hong C H, Lee J C, Wu C H, Der Danielian M, Wu V, Newman R, Lichtenstein A K, Berenson J R. J Immunol. 1995;155:2487–2497. [PubMed] [Google Scholar]
- 35.Sahota S S, Leo R, Hamblin T J, Stevenson F K. Blood. 1996;87:746–755. [PubMed] [Google Scholar]
- 36.Takahashi M, Tsukada T, Kojima M, Koide T, Koike T, Takahashi H, Sakai C, Kashimura M, Shibata A. Blood. 1986;67:1710–1713. [PubMed] [Google Scholar]
- 37.Klein S, Radbruch A. Cell Immunol. 1994;157:106–117. doi: 10.1006/cimm.1994.1209. [DOI] [PubMed] [Google Scholar]
- 38.Spira G, Gregor P, Aguila H L, Scharff M D. Proc Natl Acad Sci USA. 1994;91:3423–3427. doi: 10.1073/pnas.91.8.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams M E, Westermann C D, Swerdlow S H. Blood. 1990;76:1387–1391. [PubMed] [Google Scholar]
- 40.Tsujimoto Y, Finger L R, Yunis J, Nowell P C, Croce C M. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 41.Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P. Proc Natl Acad Sci USA. 1982;79:7837–7841. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen-Ong G L, Keath E J, Piccoli S P, Cole M D. Cell. 1982;31:443–452. doi: 10.1016/0092-8674(82)90137-4. [DOI] [PubMed] [Google Scholar]
- 43.Ye B H, Rao P H, Chaganti R S, Dalla-Favera R. Cancer Res. 1993;53:2732–2735. [PubMed] [Google Scholar]
- 44.Kobayashi H, Espinosa R, III, Thirman M J, Gill H J, Fernald A A, Diaz M O, Le Beau M M, Rowley J D. Blood. 1993;82:547–551. [PubMed] [Google Scholar]
- 45.Weh H J, Bartl R, Seeger D, Selbach J, Kuse R, Hossfeld D K. Leukemia. 1995;9:2119–2122. [PubMed] [Google Scholar]
- 46.Tricot G, Barlogie B, Jagannath S, Bracy D, Matter S, Vesole D H, Naucke S, Sawyer J R. Blood. 1995;86:4250–4256. [PubMed] [Google Scholar]