

Abstract
The 90-kDa heat shock protein (Hsp90) is a molecular chaperone that is very abundant even at normal temperature. It is highly conserved and essential for viability in yeast. To delineate functional domains of Hsp90, we have performed a deletion analysis of one of the two Hsp90 isoforms from budding yeast, Hsp82. The Hsp82 derivatives were tested for complementation of a Hsp90-deficient yeast strain and for their ability to function in two signal transduction pathways that depend on Hsp90. Surprisingly, we found that two salient features of Hsp90 sequences from eukaryotes, the N-terminal charged domain and the extremely conserved C-terminal pentapeptide MEEVD, are dispensable for viability as well as for proper regulation of vertebrate steroid receptors and for pheromone signaling. Moreover, we describe, to our knowledge, the first dominant negative mutant of Hsp90: A Hsp82 derivative that lacks amino acids 538–552 fails to complement but has a dominant negative effect on viability of wild-type strains at moderately elevated temperatures. This mutant may become a valuable tool to study Hsp90 functions both in yeast and in mammalian cells.
Keywords: heat shock protein 90, molecular chaperone, pheromone signaling, steroid receptors, dominant negative mutant
The 90-kDa heat shock protein (Hsp90) (for reviews, see refs. 1 and 2) is a ubiquitous and abundantly expressed cytosolic protein even under normal growth conditions. It is highly conserved from bacteria to mammals. Hsp90 can act as a molecular chaperone in vitro to promote refolding of denatured proteins (refs. 3 and 4; see also refs. 5 and 6) and to prevent protein unfolding and aggregation (4, 7, 8, 9). Two genes encode closely related isoforms in mammals as well as in the budding yeast Saccharomyces cerevisiae. Deletion experiments in yeast show that the expression of at least one of the two Hsp90 isoforms, either Hsp82 or Hsc82, is essential for viability (10). Interestingly, the Escherichia coli homologue of Hsp90, HtpG, appears to be dispensable (11). Several studies have revealed that Hsp90 interacts either stably or transiently with various proteins but the precise functions of Hsp90 in these complexes remain unclear (ref. 1; see also ref. 12).
The interaction of Hsp90 with steroid receptors has been extensively investigated. A variety of in vitro and in vivo studies have revealed that steroid receptors are complexed with Hsp90 and several other proteins in the absence of hormone (see for example refs. 12, 13, 14, 15, 16, 17). The interaction between Hsp90 and steroid receptors is established via their hormone binding domain (HBD) (18, 19). Upon ligand binding, the HBD undergoes a conformational change (20, 21) and Hsp90 is released. Steroid receptors and many heterologous proteins fused to the HBD are maintained inactive in the absence of hormone. We have therefore hypothesized that the hormone-reversible inactivation function of the HBD is mediated by Hsp90 possibly by steric hindrance (22, 23, 24). Further insights into the role of Hsp90 in the regulation of this particular signal transduction pathway come from studies made in yeast. Vertebrate steroid receptors expressed in yeast strains with low-level (ref. 25; see also ref. 26) or specific point (27, 28, 29) mutants of Hsp82 show a defective hormonal response that, in the case of the glucocorticoid receptor, has been shown to be due to a decrease in the ligand binding affinity (28). Thus, Hsp90 may have a dual role: It ensures that receptors are kept inactive in the absence of hormone and helps them to respond specifically and efficiently to ligand. Recently, we have also found that Hsp90 is required for the yeast pheromone signaling pathway (J.-F. L. and D.P., unpublished results). Both the induced and the basal activities of the pathway are defective in yeast strains expressing low-level or specific point mutations of Hsp82 or human Hsp90β.
A large diversity of HSP90 variants, which complement yeast strains carrying disruptions of the essential HSP90 genes, have been described (10, 25, 27, 28, 29, 30, 31, 32, 33). This includes point mutations and homologues from other species, but an attempt at delimiting essential domains for viability with a deletion analysis in a homologous system has not been made. In vitro assembly assays between Hsp90 derivatives and the progesterone receptor (PR) (34) and in vivo complex formation between Hsp90 mutants and several steroid receptors upon coexpression in insect and mammalian cells (33, 35, 36) have highlighted several regions in Hsp90 that are important for this particular interaction. In this work, we have carried out a mutational analysis of HSP82. We have examined in vivo the phenotypes of the deletion mutants with respect to (i) the complementation of a Hsp90-defective yeast strain for viability and (ii) specific Hsp90 functions in the regulation of vertebrate steroid receptors and yeast pheromone signaling.
MATERIALS AND METHODS
Plasmids.
The plasmid pTT8 contains the HSP82 sequence under the control of the GAL1 promoter, the TRP1 marker, and a CEN/ARS replicon (25). The expression vectors p2U (32) and p2HG (25) contain the URA3 and HIS3 markers, respectively, the constitutive glyceraldehyde-3-phosphate dehydrogenase promoter, and a 2-μm replicon.
All constructions containing HSP82 sequences were made with fragments excised enzymatically or by PCR from plasmid pTT8 and initially introduced into p2U. To construct p2U/Hsp82, HSP82 sequences were introduced as a BamHI–SacI fragment. p2U/Hsp82(1–704) was obtained by introducing a stop codon after codon 704 by PCR. p2U/Hsp82(1–685) was generated by the deletion of a AlwNI–SacI fragment from p2U/Hsp82 that results in the addition of the 5 amino acids LLFAL after Glu-685. p2U/Hsp82(1–652) was obtained by introducing a stop codon after codon 652 by subcloning a XbaI–HincII fragment from pTT8 into a pUC18 derivative containing a stop codon in the proper reading frame. p2U/Hsp82(Δ11-107) was made by ligating the AluI sites located in codons 9–10 and codons 106–107. The deletion of p2U/Hsp82(Δ211-259) was created by PCR; the resulting amino acid sequence at the deletion reads Thr-210–Met–Val-260. To generate p2U/Hsp82(Δ319-353), AluI restriction sites located in codons 318–319 and 353–354 were joined. p2U/Hsp82(Δ538-552) was obtained by fusing the AluI restriction site located in codons 537–538 to the StuI site located in codons 552–554. p2U/Hsp82(Δ582-601) was created by introducing a BamHI site by PCR at the deletion, thereby joining codons 581 and 602.
Plasmids p2HG/Hsp82, p2HG/Hsp82(1–704), p2HG/Hsp82(1–685), and p2HG/Hsp82(Δ211-259) were obtained by excising the HSP82 sequences contained in BamHI–SacI fragments from the p2U plasmids and introducing them into p2HG.
The derivatives of plasmid p2U/Hsp82(Δ538-552) with a deletion of amino acids 1–354 all contain the hemagglutinin (HA) epitope (YPYDVPDYA) at the N terminus. Plasmids p2U/fluHsp82(Δ1-354/Δ538-552Δ/704-709), p2U/fluHsp82(Δ1-354/Δ538-552/Δ685-709), and p2U/fluHsp82(Δ1-354/Δ538-552/Δ652-709) were obtained by combining the HSP82 sequences from p2U/fluHsp82(Δ1-354) and p2U/Hsp82(Δ538-552) with the HSP82 sequences from p2U/Hsp82(1–704), p2U/Hsp82(1–685), and p2U/Hsp82(1–652), respectively.
The human estrogen receptor (hER) expression vector pG/hER was made by cloning the entire hER coding sequence from p2HG/hER (25) into the expression vector pG-1 (37). The reporter plasmid with glucocorticoid response elements pUCΔSS-26X is a pUC derivative of plasmid pSX26.1 (38). pUCΔSS-ERE is a reporter plasmid containing estrogen response elements (25). The rat glucocorticoid receptor (rGR) and the human progesterone receptor (hPR) were expressed from plasmids pG/N795 (38) and pYE10hPR1A (a gift from H. Gronemeyer and P. Chambon, Institut de Génétiqùe et de Biologie Moléculaire et Cellulaire, Illkirch, France), respectively.
Strains and Complementation Assay.
The strain HH1-KAT6 (MATα ade2 leu2 ura3 his3 trp1 hsc82::LEU2 hsp82::LEU2/HSP90β-CEN/ARS-HIS3 [pGal1-hhsp90]), which has been described (32), was used to test the viability of HSP82 deletion mutations. The strain was transformed by the lithium acetate/polyethylene glycol method. For complementation assays, transformants were cultured with galactose as the carbon source and then streaked onto galactose or glucose plates. GRS4 Mata was obtained as follows: GRS4 (25), which is MATα, was transformed with plasmid pHO encoding the S. cerevisiae HO endonuclease under the control of its own promoter (a gift from M. Belin-Collart, University of Geneva, Geneva). Diploids were subsequently sporulated and individual spores were characterized after tetrad dissection. The HH1a series of strains HH1a-p2HG/Hsp82, HH1a-p2HG/Hsp82(1–704), HH1a-p2HG/Hsp82(1–685), and HH1a-p2HG/Hsp82(Δ211-259), expressing the wild-type HSP82 and the HSP82 deletions mutations, were derived from GRS4 Mata by plasmid shuffling with the corresponding p2HG expression vectors.
β-Galactosidase Assays.
The HH1a strains HH1a-p2HG/Hsp82, HH1a-p2HG/Hsp82(1–704), HH1a-p2HG/Hsp82(1–685), and HH1a-p2HG/Hsp82(Δ211-259) were transformed with plasmids encoding the mammalian steroid receptors along with their corresponding β-galactosidase reporter genes. pUCΔSS-26X was used to monitor the activation of the rGR and the hPR receptors. pUCΔSS-ERE served to assess the activation of the hER. Transformants cultured overnight at 30°C in selective medium containing 2% glucose as the carbon source were diluted 1:20 to obtain low-density cultures and incubated for 12 h in the presence of hormone (1000× stocks in ethanol) or ethanol alone. β-Galactosidase assays were performed as described (39) and corrected for cell density. Deacylcortivazol (DAC) was obtained from Roussel-Uclaf.
Pheromone Signaling Assays.
The control strain E929-6C-24 (relevant genotype: MATa Δste11) (a gift from B. Errede, University of North Carolina, Chapel Hill) and the HH1a strains HH1a-p2HG/Hsp82, HH1a-p2HG/Hsp82(1–704), HH1a-p2HG/Hsp82(1–685), and HH1a-p2HG/Hsp82(Δ211-259) were precultured in YEPD, diluted to a density of 1.2 × 107 cells per ml, streaked onto YEPD plates containing 10 mM sodium citrate (pH 4.3) with or without 5 μM α-factor (Bachem), and incubated at 30°C for 3 days.
Preparation of Cell Extracts and Immunoblot Analysis.
Cell extracts were prepared at 4°C by breaking the cells with glass beads in 10 mM Tris·HCl, pH 7.5/50 mM NaCl/1 mM DTT/20 mM sodium molybdate/15 mM MgCl2/10% glycerol/1 mM phenylmethylsulfonyl fluoride (PMSF)/protease inhibitors (aprotinin, each at 0.5 μg/μl/leupeptin/pepstatin A). Samples were frozen in liquid nitrogen and stored at −70°C. Extracts, 10 μg of each, as determined with the Bio-Rad Bradford reagent, were boiled in SDS/sample buffer for 5 min and loaded onto a SDS/7.5% polyacrylamide gel. Proteins were transferred to nitrocellulose. The blot was blocked with Tris-buffered saline/0.05% Tween 20 (TBST) containing 5% milk powder and probed with a chicken antiserum raised against recombinant yeast Hsp82 in TBST/5% milk powder at room temperature for 1 h. The membrane was washed for three 10-min periods with TBST. The secondary antibody was alkaline phosphatase-conjugated goat anti-chicken IgY (Promega) and was used in TBST/5% milk powder at room temperature for 1 h. After three washes with TBST, the blot was developed with the nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate reagent for alkaline phosphatase.
RESULTS
Analysis of the Viability Function of HSP82 Deletion Mutations.
To test deletion mutations of HSP82, we adopted the strategy depicted in Fig. 1A. Plasmids encoding constitutively expressed Hsp82 derivatives (as shown in Fig. 1B) were introduced into a yeast strain that expresses the human HSP90β (hHSP90β), under the control of the conditional GAL1 promoter, instead of the chromosomal HSP82 and HSC82. Individual transformants were cultured in selective medium containing galactose as the carbon source and streaked onto glucose or galactose plates. Only transformants with viable HSP82 deletion mutations can grow on plates with glucose that represses the expression of hHSP90β. To avoid missing temperature-sensitive mutations, transformants were tested at 21°C, 30°C, and 37°C.
Figure 1.
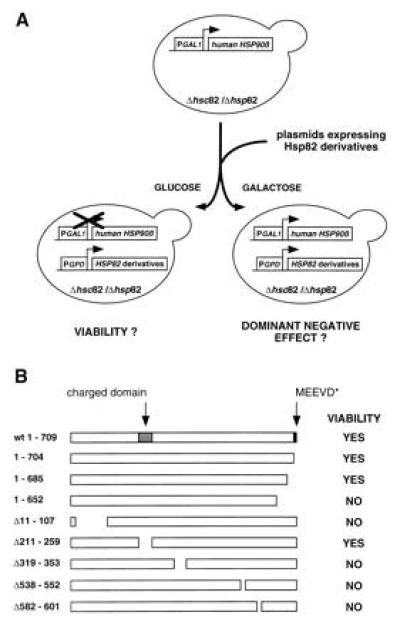
Viability of HSP82 deletion mutations. (A) Strategy to analyze the viability function of Hsp82 deletion mutants. Plasmids encoding Hsp82 derivative were introduced into a S. cerevisiae strain with a deletion of both HSP90 genes, HSP82 and HSC82. Viability is ensured by the expression of the human HSP90β under the control of the conditional GAL1 promoter. Transformants were streaked onto galactose or glucose plates, which resulted in the expression or repression of hHSP90β, respectively. Transformants with viable HSP82 deletion mutations are able to form colonies on glucose plates whereas those with dominant negative HSP82 deletion mutants fail to form colonies on galactose plates. (B) Schematic representation of the HSP82 deletion mutations and their complementing activities. The positions of two eukaryote-specific regions, the charged domain, and the C-terminal MEEVD motif are indicated.
In contrast to many other proteins, Hsp82 tolerates very few truncations. Most of the deletions yielded nonviable derivatives at all temperatures tested. An immunoblot experiment confirmed that all Hsp82 mutants are expressed (data not shown; see also below). The three Hsp82 derivatives Hsp82-(1–704), Hsp82-(1–685), and Hsp82-(Δ211-259) were found to be viable at all temperatures tested (Fig. 1B). Remarkably, this indicates that both sequences that are highly characteristic for eukaryotes, the charged domain in the N-terminal third and the C-terminal pentapeptide MEEVD, are dispensable. All eukaryotic Hsp90 proteins contain a highly charged domain in the N-terminal third of the protein. In the yeast S. cerevisiae, it is located between amino acid 211 and 259 and contains the sequence N3K2. The C-terminal pentapeptide MEEVD is identical in all known eukaryotic sequences except for one amino acid change in some trypanosomatids. These sequences are absent from HtpG, the E. coli homologue of Hsp90, that is not essential for viability in bacteria (11). It has previously been reported that the truncation of the C-terminal 49 amino acids of human Hsp90α abolishes homodimerization and complementation in yeast (30). Our results show that the removal of the last 24 amino acids from Hsp82-(1–685) has no effect on viability, thus narrowing down the C-terminal end point required for this essential Hsp90 function. To address the possibility that Hsp82 contains functional domains that can function in an independent fashion in trans, we tested whether the coexpression of distinct nonviable deletion mutations could restore viability. This does not appear to be the case since all combinations failed to complement a Hsp90 deficiency (data not shown). Taking advantage of the plasmid shuffling technique, we generated a series of isogenic strains expressing the wild-type Hsp82, Hsp82-(1–704), Hsp82-(1–685), or Hsp82-(Δ211-259). When compared with wild-type Hsp82, all Hsp82 derivatives are expressed at similar levels (Fig. 2A) and all four strains have identical growth rates (Fig. 2B).
Figure 2.
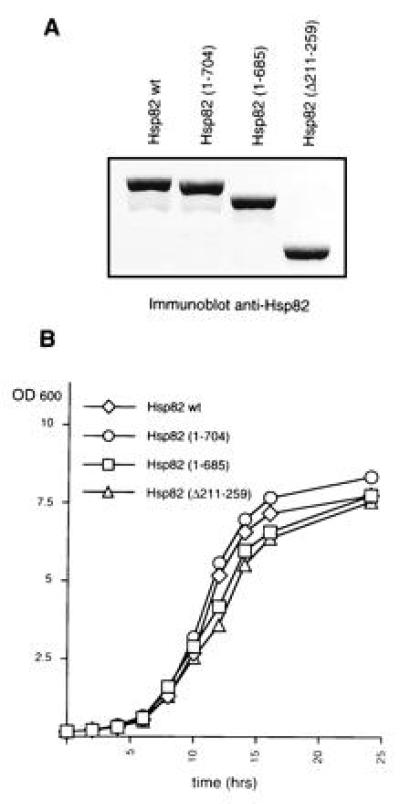
Yeast strains express HSP82 deletion mutations at similar levels and have identical growth rates. (A) Protein levels of wild-type Hsp82 and Hsp82 derivatives. The immunoblot of 10 μg of extract of each corresponding strain was probed with a chicken antiserum raised against recombinant yeast Hsp82. (B) Growth curves of strains expressing wild-type HSP82 or HSP82 deletion mutations. Cultures were started at a cell density of 3.5 × 106 cells per ml in YEPD medium (time 0) and incubated at 30°C. Data represent the average of the OD600 from two yeast clones.
Characterization of a Dominant Negative Hsp82 Mutant.
While performing the HSP82 deletion analysis, we made a particular effort to find dominant negative Hsp82 mutants. This was done by coexpressing hHSP90β and the HSP82 deletion mutations and by testing for dominant negative effects under normal or stress conditions. As indicated in Fig. 3A, the short deletion of codons 538 to 552 [mutant Hsp82-(Δ538-552)] yields a protein that behaves as a dominant negative mutant. Cells that express this mutant fail to grow at elevated temperature, typically 38°C. Microscopic examination did not reveal any obvious phenotype at permissive (30°C) or restrictive (38°C) temperature. This toxic effect of Hsp82-(Δ538-552) was observed in strains containing only the human Hsp90β or expressing endogenous Hsp82 and/or Hsc82. Both haploid and diploid yeast strains were sensitive to the dominant negative mutant (data not shown). For further analysis, a strain carrying the endogenous HSP82 and HSC82 genes was chosen. A deletion analysis of Hsp82-(Δ538-552) revealed that additional N- and C-terminal sequences can be removed without affecting the dominant negative effect (Fig. 3A). Interestingly, the C-terminal end points are the same for complementation and for the dominant negative effect: A derivative with a truncation to amino acid 685 is still toxic, whereas further truncation to amino acid 652 abolishes the dominant negative effect. In contrast, a large portion of the N-terminal half can be removed without interfering with the dominant negative effect.
Figure 3.
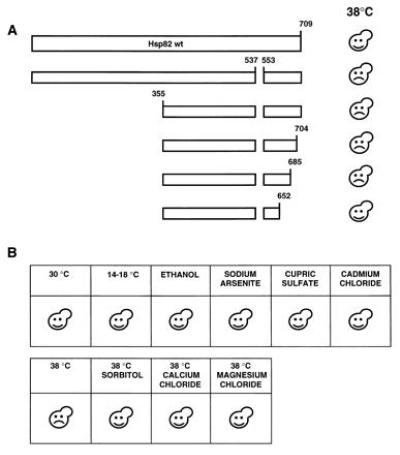
Characterization of a dominant negative Hsp82 mutant. (A) Schematic representation of a deletion analysis of the dominant negative mutant Hsp82-(Δ538-552). Plasmids encoding Hsp82 mutants were introduced into a wild-type yeast strain and transformants were streaked onto minimal plates and incubated at 30°C or 38°C. (B) The dominant negative effect is limited to heat stress. A wild-type yeast strain expressing the dominant negative Hsp82 mutant was streaked onto minimal plates. Where indicated, 6% ethanol, 0.1 mM sodium arsenite, 3 mM cupric sulfate, 10 μM cadmium chloride, 1 M sorbitol, 100 mM calcium chloride, and 100 mM magnesium chloride were added. Unless indicated, plates were incubated at 30°C. Happy and sad faces indicate that the transformants succeeded or failed, respectively, to grow.
The heat shock response and, thus, heat shock proteins are induced by many types of stress in addition to high temperature (40). Exposure to stress agents such as ethanol and heavy metals leads to protein damage that also elicits the heat shock response. As shown in Fig. 3B, the dominant negative effect of Hsp82-(Δ538-552) is only apparent upon exposure to elevated temperatures. Cells expressing Hsp82-(Δ538-552) are not more sensitive than wild-type cells to ethanol, arsenite, copper, or cadmium. Interestingly, the dominant negative effect of Hsp82-(Δ538-552) is neutralized when osmotic stabilizers such as sorbitol, CaCl2, or MgCl2 are added to the medium (Fig. 3B).
Exposing yeast cells to moderately high temperature (37°C) prepares them to survive more extreme temperatures, a phenomenon referred to as thermotolerance (see ref. 40). A possible effect of the dominant negative Hsp82 on thermotolerance was examined. However, we found that Hsp82-(Δ538-552) has no effect on the short-term survival of cells upon exposure to 50°C, with or without pretreatment at 37°C (data not shown).
The Charged and C-Terminal Domains Are Dispensable for Proper Steroid Receptor Regulation.
Genetic studies in yeast have confirmed that Hsp90 is essential for the proper regulation of the glucocorticoid (GR), estrogen (ER), progesterone (PR), mineralocorticoid, and retinoid receptors (25, 26, 27, 28, 29). We therefore assessed the effects of viable Hsp82 derivatives on the regulation of GR, ER, and PR. Isogenic strains expressing either wild-type Hsp82 or Hsp82 mutants were transformed with plasmids encoding the mammalian steroid receptors and a β-galactosidase reporter gene under the control of a minimal promoter linked to the appropriate hormone response element. As shown in Fig. 4, the hormonal dose–response curves obtained with the strains expressing Hsp82 mutants are similar to those obtained with the strain expressing wild-type Hsp82 or with a wild-type strain with the normal chromosomal HSP82 and HSC82 genes. In some cases, the C-terminal truncations even lead to a higher maximal response, but the hormone concentrations required for half-maximal response are similar.
Figure 4.
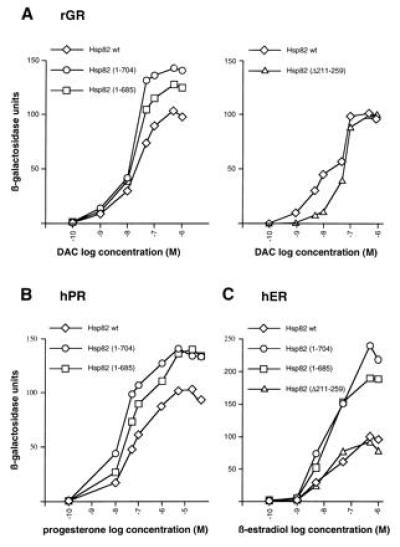
Regulation of steroid receptors in strains with Hsp82 derivatives. Strains were cotransformed with expression vectors for the rat GR (rGR) (A), human PR (hPR) (B), and human ER (hER) (C) and the appropriate β-galactosidase reporter gene (pUCΔSS-26X for rGR and hPR; pUCΔSS-ERE for hER). The two panels for rGR represent two separate sets of experiments. The hPR was not tested with Hsp82-(Δ211-259). Hormones were added to low-density cultures from 1000-fold stocks to the indicated final concentrations (logarithmic scale). Inductions were done for 12 h. β-Galactosidase units were corrected for cell density. Data represent the average of the β-galactosidase activities from two yeast clones. DAC, deacylcortivazol.
The Charged and C-Terminal Domains Are Dispensable for Pheromone Signaling.
Pheromone signaling is defective in yeast strains expressing low-level or specific point mutations of Hsp82 or human Hsp90β (J.-F.L. and D.P., unpublished results). To determine if the two Hsp82 regions, which are dispensable for viability, are required for this Hsp90 function, strains with viable HSP82 deletions were tested for their ability to arrest growth in response to pheromone. As shown in Fig. 5, all strains were growth-arrested upon exposure to pheromone, indicating that the same two domains are also dispensable for pheromone signaling. Moreover, the deletion mutations of HSP82 that fail to complement a defective yeast strain for viability also fail to rescue the pheromone pathway in a yeast strain living on human Hsp90β (data not shown).
Figure 5.
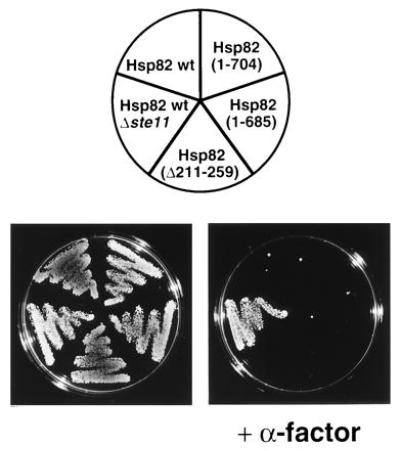
Pheromone signaling in strains with Hsp82 derivatives. Strains precultured in YEPD were diluted to a density of 1.2 × 107 cells per ml and streaked onto YEPD plates with and without 5 μM α-factor. The control strain E929-6C-24 (Hsp82 wt, Δste11) is not growth arrested upon exposure to α-factor since it lacks the kinase Ste11, an essential component of the pheromone signaling pathway. The plates were photographed after 3 days of incubation at 30°C.
DISCUSSION
Viability of HSP82 Deletion Mutations.
We have carried out a mutational analysis of Hsp82, one of the two yeast homologues of Hsp90. The mutants were examined with respect to the general role of Hsp90 for viability and to two specific nonessential functions: the regulation of vertebrate steroid receptors and of pheromone signaling. We found that the viability function of Hsp82 does not tolerate extensive truncations, indicating that several domains are required to ensure viability or that major alterations destabilize the structure of the protein.
Surprisingly, two salient features of eukaryotic Hsp90 molecules, the charged domain [mutant Hsp82-(Δ211-259)] and the C-terminal pentapeptide MEEVD [mutant Hsp82-(Δ1-704)], are dispensable for viability. Remarkably, Hsp90 shares the very last 4 amino acids, EEVD, with Hsp70. Biochemical studies have shown that the deletion or the mutation of the conserved EEVD motif in Hsp70 reduce both substrate binding and refolding of denatured substrate proteins (41). Our results indicate that the MEEVD motif of Hsp90 may not have the same function as the EEVD motif of Hsp70 or that this function is not required for viability. Since both regions are dispensable for viability the significance of this high conservation in Hsp90 remains unclear although it is conceivable that they are involved in specific nonessential functions of Hsp90 that have yet to be discovered (see also below). The E. coli Hsp90 homologue HtpG shares 42% sequence identity with yeast Hsp82, but it lacks the extreme N-terminal portion, the charged domain, and the pentapeptide MEEVD. We have shown (32) that HtpG fails to complement a Hsp90 defective yeast strain. Our results indicate that the failure of the bacterial counterpart to complement yeast is not solely due to the absence of the charged domain and the MEEVD motif. Indeed, several HtpG–Hsp82 chimeras are also unable to fulfill the requirements for viability (G. Palmer and D.P., unpublished results).
Several point mutations of HSP82 have been isolated that confer a temperature-sensitive growth phenotype (27, 29, 31). However, none of them is located in regions that we have found to be dispensable for viability. HSP82 point mutations that totally abrogate the viability function have not been reported, but none of the previous studies had specifically looked for them.
To date, very few functional sites/domains of Hsp90 have been mapped. The 49 C-terminal residues of human Hsp90α are required for dimerization and viability (30). Our viable mutant Hsp82-(1–685) further narrows down the C-terminal end point required for viability. The sequence of amino acids 500–520 of mouse Hsp90α (Hsp86) has been reported to be a calmodulin-binding domain (42) and the sequences of amino acid 363–373, 513–524, and 534–549 of mouse Hsp90α have been proposed to form an ATP binding domain (43). The removal of amino acid 319–353 [mutant Hsp82-(Δ319-353)], which yields a noncomplementing molecule, would destroy the proposed ATP binding site. However, ATP binding and ATPase activity of Hsp90 remain controversial (1). A detailed biochemical comparison of wild-type Hsp82 and Hsp82-(Δ319-353) might help to elucidate the significance of these activities for Hsp90 function.
A Dominant Negative Hsp82 Mutant.
Of all the noncomplementing mutants, the first dominant negative Hsp82 mutant [Hsp82-(Δ538-552)] to be reported, is the most remarkable one. Its toxic effect is only apparent upon exposure to elevated temperatures and is neutralized by osmotic stabilizers. Note that, at permissive temperature, growth rates are only marginally affected and steroid receptor signaling is unperturbed (data not shown). At restrictive temperature, Hsp82-(Δ538-552) might be toxic by heterodimerization with wild-type Hsp82 and Hsc82 or by nonproductive interactions with target or regulator proteins. Alternatively, if it were itself heat-labile, it could poison cells at elevated temperature by increasing the burden of denaturated proteins. According to this view, hyperosmotic conditions could stabilize the temperature-sensitive Hsp82-(Δ538-552) molecule. However, we favor the hypothesis that the dominant negative Hsp82 mutant interferes with Hsp90 functions by interacting with target and/or regulator proteins in a nonproductive fashion. This is supported by two observations: (i) a deletion analysis of Hsp82-(Δ538-552) showed that the C-terminal Hsp82 sequences required for the dominant negative phenotype match those necessary for viability and (ii) human Hsp90β functions in yeast are also blocked by the mutant Hsp82-(Δ538-552). It is unlikely that Hsp82-(Δ538-552) would form heterodimers with Hsp82, Hsc82, and human Hsp90β since mixed heterodimers are not even formed by the two endogenous isoforms present in mammalian cells (44).
The characterization of second-site suppressors might help to clarify the mode of action of the dominant negative Hsp82 mutant and to identify essential Hsp90 interaction partners. As Hsp90 is ubiquitously and abundantly expressed, technical problems usually render in vivo studies on this molecular chaperone difficult in mammalian cells. Given that the dominant negative Hsp82 mutant works in yeast cells expressing human Hsp90β, it might provide a valuable tool to interfere with Hsp90 functions in mammalian cells.
Domains of Hsp82 for Signal Transduction.
We have further contributed to delimiting the essential regions of Hsp90 in signal transduction by steroid receptors and pheromones in an in vivo setting with physiological concentrations of Hsp82 derivatives. The strength of our yeast system is that it provides very strong genetic data on the requirement for certain protein domains. With the current strategy, its weakness is that only viable HSP82 mutations can be further analyzed for specific functions of Hsp90 in cellular processes that are not essential such as signal transduction.
Other studies had previously attempted to map Hsp90 domains that are necessary for the formation of functional complexes with steroid receptors in vitro or upon massive overexpression in cells that also contained wild-type Hsp90. Sullivan and Toft (34) demonstrated that in vitro-translated Hsp90 can form a functional complex with the chicken PR in a process requiring Hsp70, ATP hydrolysis, and additional factors provided by the reticulocyte lysate. This assay was exploited to map the domains of the chicken Hsp90 that are required for functional interaction with PR (34). They found that the N-terminal half, which includes the charged domain, and the MEEVD motif are not essential for complex formation. While this is in perfect agreement with our in vivo results, two other studies, addressing the role of the charged domain of Hsp90 for the regulation of steroid receptors, have provided conflicting data: (i) A synthetic peptide corresponding to the charged domain and an antibody directed against this peptide have been shown to destabilize Hsp90–GR complexes in vitro (45). However, control peptides with similar charge were not tested. (ii) A large deletion encompassing the sequences encoding the charged domain in the chicken Hsp90 has been reported to abolish complex formation with GR upon coexpression in the baculovirus system (35). We consider this particular mutation as a loss-of-function mutation since it extended considerably beyond the charged domain. A slightly smaller deletion mutant of chicken Hsp90 has very recently been reported to be capable of complementation in yeast and interaction with ER in mammalian COS cells (33).
Several other deletions in the sequences encoding the C-terminal half of chicken Hsp90 have been shown to interfere with proper complex formation with steroid receptors (33, 34, 35). Of particular interest is the mutant Δ601-620 in chicken Hsp90 (34), which corresponds to our mutant Hsp82-(Δ582-601) in yeast Hsp82. This mutation completely abolished the formation of complexes with PR in vitro (34) and with ER in cotransfected COS cells (33). In yeast, the equivalent mutant was not viable, indicating that these 20 amino acids are also essential for viability.
The power of the yeast system could be considerably increased if nonviable Hsp90 mutants could be tested for other Hsp90 functions. Although we have found that the viability function of Hsp82 and its function in pheromone signaling could not be provided in trans by two separate molecules, a similar approach might work for studying steroid receptor signaling. Viability could be ensured by one of the Hsp82 mutants that is defective for steroid receptor signaling (25, 27, 29). One could then attempt to complement this particular defect with a series of derivatives of yeast or vertebrate Hsp90.
Acknowledgments
We thank S. Lindquist, H. Gronemeyer, P. Chambon, B. Errede, M. Belin-Collart, and H. Bussey for plasmids and strains. We are grateful to Katharina Strub and Holly Goodson for critical comments on the manuscript. This work was supported by the Swiss National Science Foundation and the Canton de Genève.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: HBD, hormone binding domain; PR, progesterone receptor; ER, estrogen receptor; GR, glucocorticoid receptor; h, human; r, rat.
References
- 1.Jakob U, Buchner J. Trends Biochem Sci. 1994;19:205–211. doi: 10.1016/0968-0004(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 2.Georgopoulos C, Welch W J. Annu Rev Cell Biol. 1993;9:601–634. doi: 10.1146/annurev.cb.09.110193.003125. [DOI] [PubMed] [Google Scholar]
- 3.Wiech H, Buchner J, Zimmermann R, Jakob U. Nature (London) 1992;358:169–170. doi: 10.1038/358169a0. [DOI] [PubMed] [Google Scholar]
- 4.Yonehara M, Minami Y, Kawata Y, Nagai J, Yahara I. J Biol Chem. 1996;271:2641–2645. doi: 10.1074/jbc.271.5.2641. [DOI] [PubMed] [Google Scholar]
- 5.Shaknovich R, Shue G, Kohtz D S. Mol Cell Biol. 1992;12:5059–5068. doi: 10.1128/mcb.12.11.5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shue G L, Kohtz D S. J Biol Chem. 1994;269:2707–2711. [PubMed] [Google Scholar]
- 7.Miyata Y, Yahara I. J Biol Chem. 1992;267:7042–7047. [PubMed] [Google Scholar]
- 8.Jakob U, Lilie H, Meyer I, Buchner J. J Biol Chem. 1995;270:7288–7294. doi: 10.1074/jbc.270.13.7288. [DOI] [PubMed] [Google Scholar]
- 9.Jakob U, Meyer I, Bugl H, Andre S, Bardwell J C, Buchner J. J Biol Chem. 1995;270:14412–14419. doi: 10.1074/jbc.270.24.14412. [DOI] [PubMed] [Google Scholar]
- 10.Borkovich K A, Farrelly F W, Finkelstein D B, Taulien J, Lindquist S. Mol Cell Biol. 1989;9:3919–3930. doi: 10.1128/mcb.9.9.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bardwell J C A, Craig E A. J Bacteriol. 1988;7:2977–2983. doi: 10.1128/jb.170.7.2977-2983.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith D F, Toft D O. Mol Endocrinol. 1993;7:4–11. doi: 10.1210/mend.7.1.8446107. [DOI] [PubMed] [Google Scholar]
- 13.Pratt W B. Mol Cell Endocrinol. 1990;74:C69–C76. doi: 10.1016/0303-7207(90)90198-h. [DOI] [PubMed] [Google Scholar]
- 14.Pratt W B. J Biol Chem. 1993;268:21455–21458. [PubMed] [Google Scholar]
- 15.Rexin M, Busch W, Segnitz B, Gehring U. J Biol Chem. 1992;267:9619–9621. [PubMed] [Google Scholar]
- 16.Kang K I, Devin J, Cadepond F, Jibard N, Guiocho-Mantel A, Baulieu E-E, Catelli M G. Proc Natl Acad Sci USA. 1994;91:340–344. doi: 10.1073/pnas.91.1.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segnitz B, Gehring U. Proc Natl Acad Sci USA. 1995;92:2179–2183. doi: 10.1073/pnas.92.6.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scherrer L C, Picard D, Massa E, Harmon J M, Simons S S, Jr, Yamamoto K R, Pratt W B. Biochemistry. 1993;32:5381–5386. doi: 10.1021/bi00071a013. [DOI] [PubMed] [Google Scholar]
- 19.Pratt W B, Jolly D J, Pratt D V, Hollenberg S M, Giguère V, Cadepond F M, Schweizer-Groyer G, Catelli M-G, Evans R M, Baulieu E-E. J Biol Chem. 1988;263:267–273. [PubMed] [Google Scholar]
- 20.Fritsch M, Leary C M, Furlow J D, Ahrens H, Schuh T J, Mueller G C, Gorski J. Biochemistry. 1992;31:5303–5311. doi: 10.1021/bi00138a009. [DOI] [PubMed] [Google Scholar]
- 21.Beekman J M, Allan G F, Tsai S Y, Tsai M-J, O’Malley B W. Mol Endocrinol. 1993;7:1266–1274. doi: 10.1210/mend.7.10.8264659. [DOI] [PubMed] [Google Scholar]
- 22.Picard D. Trends Cell Biol. 1993;3:278–280. doi: 10.1016/0962-8924(93)90057-8. [DOI] [PubMed] [Google Scholar]
- 23.Picard D. Curr Opin Biotechnol. 1994;5:511–515. doi: 10.1016/0958-1669(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 24.Mattioni T, Louvion J-F, Picard D. Methods Cell Biol. 1994;43:335–352. doi: 10.1016/s0091-679x(08)60611-1. [DOI] [PubMed] [Google Scholar]
- 25.Picard D, Khursheed B, Garabedian M J, Fortin M G, Lindquist S, Yamamoto K R. Nature (London) 1990;348:166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- 26.Holley S J, Yamamoto K R. Mol Biol Cell. 1995;6:1833–1842. doi: 10.1091/mbc.6.12.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bohen S P, Yamamoto K R. Proc Natl Acad Sci USA. 1993;90:11424–11428. doi: 10.1073/pnas.90.23.11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bohen S P. J Biol Chem. 1995;270:29433–29438. doi: 10.1074/jbc.270.49.29433. [DOI] [PubMed] [Google Scholar]
- 29.Nathan D F, Lindquist S. Mol Cell Biol. 1995;15:3917–3925. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minami Y, Kimura Y, Kawasaki H, Suzuki K, Yahara I. Mol Cell Biol. 1994;14:1459–1464. doi: 10.1128/mcb.14.2.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kimura Y, Matsumoto S, Yahara I. Mol Gen Genet. 1994;242:517–527. doi: 10.1007/BF00285275. [DOI] [PubMed] [Google Scholar]
- 32.Palmer G, Louvion J-F, Tibbetts R S, Engman D M, Picard D. Mol Biochem Parasitol. 1995;70:199–202. doi: 10.1016/0166-6851(95)00007-n. [DOI] [PubMed] [Google Scholar]
- 33.Meng X, Devin J, Sullivan W P, Toft D, Baulieu E-E, Catelli M-G. J Cell Sci. 1996;109:1677–1687. doi: 10.1242/jcs.109.7.1677. [DOI] [PubMed] [Google Scholar]
- 34.Sullivan W P, Toft D O. J Biol Chem. 1993;268:20373–20379. [PubMed] [Google Scholar]
- 35.Cadepond F, Binart N, Chambraud B, Jibard N, Schweizer-Groyer G, Segard-Maurel I, Baulieu E-E. Proc Natl Acad Sci USA. 1993;90:10434–10438. doi: 10.1073/pnas.90.22.10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Binart N, Lombes M, Baulieu E-E. Biochem J. 1995;311:797–804. doi: 10.1042/bj3110797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schena M, Picard D, Yamamoto K R. Methods Enzymol. 1991;194:389–398. doi: 10.1016/0076-6879(91)94029-c. [DOI] [PubMed] [Google Scholar]
- 38.Schena M, Yamamoto K R. Science. 1988;241:965–967. doi: 10.1126/science.3043665. [DOI] [PubMed] [Google Scholar]
- 39.Yocum R R, Hanley S, West R W, Jr, Ptashne M. Mol Cell Biol. 1984;4:1985–1998. doi: 10.1128/mcb.4.10.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindquist S, Craig E A. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 41.Freeman B C, Myers M P, Schumacher R, Morimoto R I. EMBO J. 1995;14:2281–2292. doi: 10.1002/j.1460-2075.1995.tb07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minami Y, Kawasaki H, Suzuki K, Yahara I. J Biol Chem. 1993;268:9604–9610. [PubMed] [Google Scholar]
- 43.Csermely P, Kahn C R. J Biol Chem. 1991;266:4943–4950. [PubMed] [Google Scholar]
- 44.Minami Y, Kawasaki H, Miyata Y, Suzuki K, Yahara I. J Biol Chem. 1991;266:10099–10103. [PubMed] [Google Scholar]
- 45.Tbarka N, Richard-Mereau C, Formstecher P, Dautrevaux M. FEBS Lett. 1993;322:125–128. doi: 10.1016/0014-5793(93)81551-a. [DOI] [PubMed] [Google Scholar]