

Abstract
The effect of ionic strength on the conformational equilibrium between the I2 intermediate and the signaling state I2′ of the photoreceptor PYP and on the rate of recovery to the dark state were investigated by time-resolved absorption and fluorescence spectroscopy. With increasing salt concentration up to ∼600 mM, the recovery rate k3 decreases and the I2/I2′ equilibrium (K) shifts in the direction of I2′. At higher ionic strength both effects reverse. Experiments with mono-(KCl, NaBr) and divalent (MgCl2, MgSO4) salts show that the low salt effect depends on the ionic strength and not on the cation or anion species. These observations can be described over the entire ionic strength range by considering the activity coefficients of an interdomain salt bridge. At low ionic strength the activity coefficient decreases due to counterion screening whereas at high ionic strength binding of water by the salt leads to an increase in the activity coefficient. From the initial slopes of the plots of log k3 and log K versus the square root of the ionic strength, the product of the charges of the interacting groups was found to be −1.3 ± 0.2, suggesting a monovalent ion pair. The conserved salt bridge K110/E12 connecting the β-sheet of the PAS core and the N-terminal domain is a prime candidate for this ion pair. To test this hypothesis, the mutants K110A and E12A were prepared. In K110A the salt dependence of the I2/I2′ equilibrium was eliminated and of the recovery rate was greatly reduced below ∼600 mM. Moreover, at low salt the recovery rate was six times slower than in wild-type. In E12A significant salt dependence remained, which is attributed to the formation of a novel salt bridge between K110 and E9. At high salt reversal occurs in both mutants suggesting that salting out stabilizes the more compact I2 structure. However, chaotropic anions like SCN shift the I2/I2′ equilibrium toward the partially unfolded I2′ form. The salt linkage K110/E12 stabilizes the photoreceptor in the inactive state in the dark and is broken in the light-induced formation of the signaling state, allowing the N-terminal domain to detach from the β-scaffold PAS core.
INTRODUCTION
PYP is a small cytoplasmic photoreceptor (14 kDa) from Halorhodospira halophila that is the structural prototype of the PAS domain family (1,2). Halorhodospira halophila is negatively phototactic in response to intense blue light with an action spectrum suggestive of PYP. The blue light absorption of PYP (λmax = 446 nm) is due to its 4-hydroxycinnamoyl chromophore that is covalently bound via a thioester linkage to Cys-69. In the initial dark state the 7–8 double bond of the chromophore is trans and the chromophore is deprotonated. Rapid photoisomerization to the cis state (3) is followed by a cascade of dark reactions between various thermal intermediates leading back to the initial state (4). The main features of the photocycle of PYP, including absorption maxima, lifetimes, protonation changes, and equilibria, are shown in Fig.1 C. The first intermediate relevant for this article is I1, which absorbs at 460 nm and still has a deprotonated chromophore. I1 decays in several hundred microseconds to I2, which has a protonated chromophore and absorbs around 370 nm (5–7). I2 decays in ∼2 ms to I2′, which is further blue-shifted to ∼350 nm (5–7). Solution studies show that the formation of I2′ is associated with a global conformational change. Circular dichroism (8) and Fourier transform infrared (9,10) spectroscopy indicate a major change in secondary structure. NMR measurements suggest increased flexibility and disorder in the N-terminal domain (11,12). I2′ decays to the initial dark state P in several hundred milliseconds. This recovery transition is associated with reisomerization, chromophore deprotonation, and reversal of the structural change. It is likely that the longest lived intermediate I2′ with the largest structural change interacts transiently with a response regulator and should be identified with the signaling state. PYP is an excellent model system for studying the mechanism of PAS domain proteins, since high resolution x-ray structures are available for the dark state (13) and a number of photocycle intermediates (14,15). The solution structure has also been characterized by high resolution NMR in the dark (16) and in the light (11).
FIGURE 1.

(A) View of the backbone structure of PYP with the N-terminal domain in the dark state on the right (wheat color) and the PAS core on the left (gray color) based on Protein Data Bank file 1NWZ (56). Lys-110 and Glu-12 are labeled in green. The side chain of K110 makes a salt bridge with E12 and a hydrogen bond with the backbone carbonyl of E9. In the mutant E12A an alternate salt bridge of K110 with the carboxyl of E9 is feasible. (B) Space filling view from the same direction as in panel A, showing that the charged groups of K110 and E12 are accessible from the surface. Figures generated using the program PyMol. (C) Simplified scheme of the photocycle of PYP from Halorhodospira halophila.
The initial light-induced structural perturbation is localized in the chromophore binding pocket on one side of the central β-sheet. The global structural change appears to involve mainly the other side of the β-scaffold, in particular the N-terminal domain. This segment of residues 1–28 forms a subdomain in the PYP structure that is packed against the central β-sheet. An important unresolved question is how the signal is transmitted across the central β-sheet. Attention has been called in this connection to the potential role of the interface between these domains in the formation of the signaling state (17,18).
The latter part of the photocycle, involving the intermediates I1, I2, and I2′, has been studied extensively (4,7,19–22). It was shown that the cycle is not unidirectional but includes back reactions and equilibria between intermediates (7,20–23). Of particular interest is the equilibrium between I2 and the signaling state I2′, which involves the global conformational change. The first evidence for this dynamic equilibrium was obtained from double flash photoreversal measurements (24). This conformational equilibrium is pH dependent with a pKa of ∼6.3 with I2′ the main species above the pKa (5–7). We found that over the pH range from 4.5 to 11.0 the recovery rate is proportional to the concentration of the I2′ intermediate (7,22).
The formation of the signaling state I2′ is associated with an increase in the radius of gyration (25) and exposure of a hydrophobic surface area (26), with hydrophobic dyes binding transiently to I2′ but not to I2 (19). The NMR spectra of a photostationary mixture of I2 and I2′ in solution showed structural disorder and exchange on the millisecond timescale (11), which were attributed to partial unfolding presumably in the I2′ state (12). Thus the rate constant for the return of I2′ to the initial dark state P can be viewed as a refolding rate constant. Its temperature dependence shows distinct non-Arrhenius behavior with a change in sign of the slope in log k against 1/T plots (26). Such a temperature dependence is typical for folding reactions and can be described satisfactorily by assuming that the reaction is associated with a large change in heat capacity (27). It has also been shown that over a wide pH and temperature range, the kinetics of refolding from the acid-denatured state of PYP, with the protonated chromophore in the cis-state, are the same as that of the photocycle recovery kinetics from I2′ to P (28). Thus, there is substantial evidence that the I2 to I2′ and the I2′ to P transitions are associated with partial unfolding and refolding, respectively.
Fig. 1 A shows that the N-terminal domain of PYP is packed against the central β-sheet of the PAS core. Our working hypothesis is that, in the I2 to I2′ transition, the N-terminal domain becomes detached and disordered, exposing the central β-sheet. We postulate further, that one of the major interactions between these two domains is the salt linkage between K110 (central β-sheet) and E12 (N-terminal domain) that is broken in the formation of I2′ and reformed in the I2′ to P transition (refolding). The space filling structure of Fig. 1 B shows that in the dark state the nitrogen of K110 and the oxygens of E12 are accessible from the surface.
Our working hypothesis is based on recent work where we showed that the I1, I2, and I2′ equilibria are strongly dependent on the salt concentration in the mutant Y98Q (18). Analysis of the amplitude spectra derived from transient absorption spectroscopy indicated that salt had its primary effect on the I2/I2′ equilibrium. Because the absorption spectra of these species were not available, it was not possible to quantify these effects further. It was concluded that salt shifts the I2/I2′ equilibrium toward I2′ and that at very low salt the amount of light-induced signaling state is quite low. We argued that in the absence of salt, the salt linkage K110/E12 between the β-scaffold (K110) and the N-terminal domain (E12) keeps the photoreceptor in the inactive state (18). Salt-induced weakening of this electrostatic interaction (“ionic lock”) would allow the light-induced detachment of the N-terminal cap from the β-sheet and permit the global conformational change that is associated with the I2 to I2′ transition (18). For wild-type, preliminary experiments indicated similar salt-induced effects on the I1 and I2/I2′ equilibria but these could not be analyzed in the absence of accurate absorption spectra for the I2 and I2′ intermediates (18). In the meantime (5–7) these spectra became available allowing for the first time a quantitative analysis.
The ion pair K110/E12 (K/D in Rb. capsulatus and Rb. sphaeroides) is conserved in all known single domain PYP species, but is not present in the multidomain species Ppr and Ppd, which have enzymatic domains and photocycles that differ strongly from that of Halorhodospira halophila PYP (29,30). In LOV domain proteins a conserved K/E salt bridge keeps two domains together and may play an important role in the functional cycle (31,32).
The goals of the studies reported here were twofold. First, to show how the ionic strength affects the I2/I2′ equilibrium and the recovery rate in wild-type and to investigate whether these salt effects can be explained on the basis of existing theories. Second, to identify the postulated ion pair with the salt bridge K110/E12 by investigating the properties of the mutants K110A and E12A.
Surprisingly little quantitative work has been done concerning the effect of ionic strength on the stability and rate of formation of salt bridges that link protein domains. Our analysis may thus be of more general interest, for example, for protein folding.
MATERIALS AND METHODS
Protein production and purification
H. halophila holo-PYP was produced by the use of the biosynthetic enzymes TAL and pCL and subsequently purified from Escherichia coli BL21 (DE3) as described (33). The mutagenesis was performed as described (34).
Transient absorption spectroscopy
Time-resolved absorption spectroscopy with 10–50 ns time resolution was performed as described (35). The wild-type spectra of the I1, I2, and I2′ intermediates at pH 7 were obtained in previous work (5,7,22). The corresponding intermediate spectra of the K110A and E12A mutants were determined at KCl concentrations of 20 mM and 1 M and pH values of 5.7 and 8 (data not shown). They were essentially the same as those of wild-type. Once these spectra are known, the time courses of the intermediate concentrations may be obtained from the transient absorbance changes ΔA(λ,t) by matrix inversion of the Lambert-Beer law (7,22).
Time-resolved fluorescence measurements
The decay of the fluorescence intensity of W119 after ps-excitation at 298 nm was measured by single photon counting as described (5). Background illumination from a light emitting diode emitting at 470 nm was used to generate a photostationary mixture of P and the I2 and I2′ intermediates in the fluorescence cuvette (5). All spectroscopic measurements were performed at room temperature and in 10 mM Tris buffer.
Dependence of the recovery rate on ionic strength
As in our previous work (18) we consider that the K110/E12 salt linkage is broken in the formation of the I2′ intermediate. In the recovery reaction, this salt bridge reforms. The latter is an intramolecular reaction in which two oppositely charged groups interact to form the salt bridge. The salt dependence of the rate of this reaction can be modeled using standard concepts of physical chemistry, the primary kinetic salt effect (36,37). Two ions A and B with charges zA and zB react via a transition state X to P:
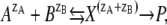 |
(1) |
in an aqueous solution of ionic strength
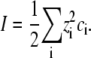 |
(2) |
The transition state X is not to be confused with the I2 intermediate. Recalling that the activities (ai) are the product of the concentrations ci and the activity coefficients fi, the rate of the reaction k is given by
 |
(3) |
where k0 is the rate constant at ionic strength zero. The ionic strength dependence of the activity coefficient fi is given by
 |
(4) |
The first term describes the electrostatic screening (Debye-Hückel). The second term describes the binding of water at high salt, which leads to a reduction in the effective concentration of water and an increase in the activity coefficient (37,38). A, B, and C are positive constants. Substitution of Eq. 4 in Eq. 3 leads to the well-known Eq. 5 (36,37):
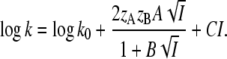 |
(5) |
The value of A at room temperature for aqueous solutions is 0.509 (37). It turns out that for our data B ≈ 0. Equation 5 thus describes a parabola in the variable 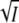 with positive curvature and a minimum. A plot of log k against
with positive curvature and a minimum. A plot of log k against 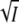 at low ionic strength allows a determination of the product zA zB of the charges that interact. In this case with K110 and E12 we expect zA zB to equal −1 and initially log k should decrease linearly with
at low ionic strength allows a determination of the product zA zB of the charges that interact. In this case with K110 and E12 we expect zA zB to equal −1 and initially log k should decrease linearly with 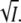 At high ionic strength the third term will dominate and log k should increase again.
At high ionic strength the third term will dominate and log k should increase again.
Effect of ionic strength on the I2/I2′ equilibrium
We consider that salt converts the “closed/locked” structure of I2 to the “open/unlocked” structure of I2′. “Closed” and “open” refer to structural states with the N-terminal domain attached to or detached from the β-scaffold, respectively. “Locked” and “unlocked” refer to the presence or absence of the K110/E12 salt linkage. The true dissociation constant K0′ for the intramolecular equilibrium between the closed form AB (I2) with fAB and the open form A+ B− (I2′) with fA, fB is given by
 |
(6) |
with the true concentrations cA = cB and cAB, where cA + cAB = c0. Introducing the normalized concentrations 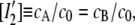 and
and 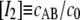 and the normalized dissociation constant K0 = K0′/c0 we get
and the normalized dissociation constant K0 = K0′/c0 we get
 |
(7) |
where F = (fA fB) / fAB. Consequently,
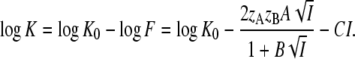 |
(8) |
Equation 8 describes a parabola in  with negative curvature and a maximum. Comparison of Eqs. 8 and 5 shows that apart from a vertical offset, these parabolas are mirror images. Note that changing the pH only affects this offset (log K0 in Eq. 8 and log k0 in Eq. 5), whereas the shape of the curves is pH independent. A plot of log K against
with negative curvature and a maximum. Comparison of Eqs. 8 and 5 shows that apart from a vertical offset, these parabolas are mirror images. Note that changing the pH only affects this offset (log K0 in Eq. 8 and log k0 in Eq. 5), whereas the shape of the curves is pH independent. A plot of log K against 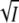 at low ionic strength allows a second independent determination of the charge product zA zB. With the sign of the expected charge product negative, the initial slope of log K against
at low ionic strength allows a second independent determination of the charge product zA zB. With the sign of the expected charge product negative, the initial slope of log K against  should be positive. We note that the initial slopes of the plots of log K and log k against
should be positive. We note that the initial slopes of the plots of log K and log k against 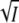 differ by a factor of −1.
differ by a factor of −1.
Effect of the I2 / I2′ equilibrium on the apparent recovery rate kapp
We showed that the intermediates I1, I2, and I2′ are in equilibrium before they decay together to P (7,18,21,22). For the recovery to P (refolding) we have
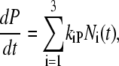 |
(9) |
where the sum over i extends over the three intermediates with time courses Ni(t). Since the equilibration between these intermediates is rapid compared to the microscopic recovery rates kiP (7,22), the intermediates decay with the same time courses
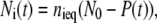 |
and Eq. 9 simplifies to
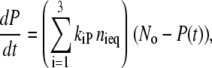 |
(10) |
where nieq is the fraction of the intermediates in the state i, and No is the total concentration of PYP. The apparent recovery rate kapp is thus given by
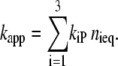 |
(11) |
From the pH dependence of the recovery rate and the fractional intermediate equilibrium concentrations, we found that from pH 4.5 to 11.0 the rate of recovery kapp is proportional to the concentration of I2′ and that the return to P occurs via I2′ (7,22). For the apparent recovery rate constant we have therefore
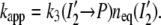 |
(12) |
We assume that this relationship (Eq. 12) between kapp and [I2′], which was derived from the pH dependence, remains valid for the salt dependence. Whereas the microscopic rate k3 was pH independent (7,22), we will find that the salt data require that k3 is salt dependent. We expect that the salt dependence of the microscopic rate k3, calculated from the experimental values of kapp and neq (I2′) according to Eq. 12, can be fitted by Eq. 5.
RESULTS
The effect of ionic strength on photocycle kinetics and intermediate populations in wild-type
Fig. 2 A shows the dependence of the photocycle kinetics on KCl at pH 7.1 at the four wavelengths 350, 390, 440, and 490 nm and four salt concentrations. These wavelengths were selected since they are diagnostic for the I2′ and I2 intermediates, the ground state depletion signal, and the I1 intermediate, respectively. Measurements were performed at the following 14 KCl concentrations: 0, 3, 6, 10, 16, 26, 41, 65, 100, 160, 260, 410, 700, and 1600 mM. Using the time traces at these four wavelengths and the known intermediate spectra (7) the time courses of the intermediate populations were calculated as described (7,22). The only assumption made is that the intermediate spectra do not depend on the ionic strength. The intermediate time courses obtained in this way at four selected salt concentrations are shown in Fig. 2 B. The traces labeled P represent the depletion and recovery of the ground state and remain almost constant in time until the recovery. The sum of the time courses of the I1, I2, I2′, and P populations is nearly zero for every ionic strength over the whole time range (not shown), showing the internal consistency of the data analysis. The results of Fig. 2 B indicate the presence of a strong salt effect on the I1, I2, and I2′ populations. In the time range where these three intermediates are in equilibrium (after the rise of I2′, ∼2 ms), the concentration of I2′ increases with increasing ionic strength at the expense of I1 and I2. Above ∼600 mM KCl, this effect is reversed. The rate constant for recovery kapp varies in the opposite direction, first decreasing and beyond 600 mM KCl increasing. We note that at 1.6 M KCl, the rise of I2′ is slower and the initial amount of I2 larger than at the lower salt concentrations, which makes for a more pronounced initial I2 peak. The dependence of K and of kapp on the ionic strength are presented in Fig. 3, A and C, respectively, for all the salt values. The equilibrium concentration values of I2 and I2′ were taken from the time courses (Fig. 2 B) at 20 ms when the three intermediates are in equilibrium. K was calculated from these values according to K = [I2′]2/[I2]. To facilitate a comparison with the theoretical predictions of Eqs. 8 and 5, log K and log k3 are plotted in Fig. 3, A and C, respectively, against the square root of the ionic strength (solid square). k3 was calculated from kapp and [I2′] using Eq. 12. The continuous curves are fits to the data according to Eqs. 8 and 5. These equations clearly provide an adequate fit over the entire ionic strength range; kapp and [I2′] are not proportional over the salt range, but by making k3 salt dependent, the relationship is retained (Eq. 12). We note that as expected from our model (Eqs. 8 and 5) the salt dependence of log K and log k3 are indeed parabolic in 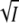 and approximate mirror images; k3 initially decreases with increasing ionic strength since the attractive interaction between the oppositely charged groups of K110 and E12 is progressively reduced by the counterion clouds.
and approximate mirror images; k3 initially decreases with increasing ionic strength since the attractive interaction between the oppositely charged groups of K110 and E12 is progressively reduced by the counterion clouds.
FIGURE 2.

(A) Time traces of the transient absorbance changes at 350, 390, 440, and 490 nm of wild-type PYP at pH 7.1 and selected KCl concentrations of 6 (solid line), 100 (dashed line), 700 (dotted line), and 1600 (dash-dotted line) mM. (B) Time courses of the I1, I2, and I2′ populations derived from the data of panel A at the corresponding salt concentrations and the intermediate spectra of Borucki et al. (7). P is the depletion of the ground state.
FIGURE 3.

(A) Plot of log K against 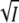 for the KCl data for wild-type of Fig. 2 (▪, transient absorbance, pH 7.1) together with the fit according to Eq. 8. The open squares are the results from the time-resolved fluorescence measurements of Fig.5 B (□, pH 8). I is the ionic strength. The results for the mutant K110A (derived from the data of Figs. 6 and 8) are marked by solid circle (transient absorbance, pH 8) and open circle (fluorescence, pH 7). To correct for the different pH values between the transient absorbance and fluorescence measurements a small offset was added to normalize the wild-type and K110A data at zero salt. (B) Plot of log K against
for the KCl data for wild-type of Fig. 2 (▪, transient absorbance, pH 7.1) together with the fit according to Eq. 8. The open squares are the results from the time-resolved fluorescence measurements of Fig.5 B (□, pH 8). I is the ionic strength. The results for the mutant K110A (derived from the data of Figs. 6 and 8) are marked by solid circle (transient absorbance, pH 8) and open circle (fluorescence, pH 7). To correct for the different pH values between the transient absorbance and fluorescence measurements a small offset was added to normalize the wild-type and K110A data at zero salt. (B) Plot of log K against 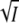 for the KCl data of E12A (•, transient absorbance, pH 8; ○, fluorescence, pH 7). The continuous curve is a fit with Eq. 8. (C) Plot of log k3 (▪) against
for the KCl data of E12A (•, transient absorbance, pH 8; ○, fluorescence, pH 7). The continuous curve is a fit with Eq. 8. (C) Plot of log k3 (▪) against 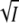 for the wild-type data of Fig. 2 together with the fit according to Eq. 5; kapp (□) is the experimental rate of recovery of the initial dark state P, and k3 was calculated from kapp and [I2′] using Eq. 12.
for the wild-type data of Fig. 2 together with the fit according to Eq. 5; kapp (□) is the experimental rate of recovery of the initial dark state P, and k3 was calculated from kapp and [I2′] using Eq. 12.
To obtain the value of the product zAzB of the interacting charges, we used the data at low ionic strength where the contributions from the C-terms can be neglected and the linear terms in 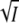 of Eqs. 5 and 8 are sufficient. The low ionic strength data for KCl (solid square), MgCl2 (solid circle), MgSO4 (solid triangle) and NaBr (star) are replotted in Fig. 4, A and B, and fitted to the linear terms in
of Eqs. 5 and 8 are sufficient. The low ionic strength data for KCl (solid square), MgCl2 (solid circle), MgSO4 (solid triangle) and NaBr (star) are replotted in Fig. 4, A and B, and fitted to the linear terms in 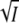 of Eqs. 5 and 8. From the common linear fits for log K and log k3 of Fig. 4, A and B, we obtained slopes of 1.6 ± 0.2 and −1.1 ± 0.1, respectively, in good agreement with the prediction of Eqs. 5 and 8 that these slopes should differ by a factor of −1. According to Eqs. 8 and 5 the slope equals −2zAzBA and +2zAzBA for the log K and log k3 plots, respectively. Using for the parameter A the value of 0.509 for aqueous solutions at room temperature (37), we obtain for the product of the charges of the reacting groups −1.5 ± 0.2 and −1.1 ± 0.1, respectively. Taking the error bars into account this result is consistent with −1, the value expected for a monovalent ion pair like K/E. The choice of 0.509 for A is reasonable, since according to Fig. 1 B the K110/E12 salt linkage is accessible from the surface in the closed/locked state and the charged side chains of K110/E12 are expected to be hydrated in the unfolded open state I2′. The linear dependence of log k3 on
of Eqs. 5 and 8. From the common linear fits for log K and log k3 of Fig. 4, A and B, we obtained slopes of 1.6 ± 0.2 and −1.1 ± 0.1, respectively, in good agreement with the prediction of Eqs. 5 and 8 that these slopes should differ by a factor of −1. According to Eqs. 8 and 5 the slope equals −2zAzBA and +2zAzBA for the log K and log k3 plots, respectively. Using for the parameter A the value of 0.509 for aqueous solutions at room temperature (37), we obtain for the product of the charges of the reacting groups −1.5 ± 0.2 and −1.1 ± 0.1, respectively. Taking the error bars into account this result is consistent with −1, the value expected for a monovalent ion pair like K/E. The choice of 0.509 for A is reasonable, since according to Fig. 1 B the K110/E12 salt linkage is accessible from the surface in the closed/locked state and the charged side chains of K110/E12 are expected to be hydrated in the unfolded open state I2′. The linear dependence of log k3 on 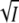 at low ionic strength is also known as the kinetic or primary salt effect (36).
at low ionic strength is also known as the kinetic or primary salt effect (36).
FIGURE 4.

(A) Fit of the linear part of the ionic strength dependence of log K (Eq. 8) for wild-type PYP. ▪, KCl; •, MgCl2; ▴, MgSO4; ★, NaBr. The data of all four salts were used in a common fit. ▵, ○, MgSO4 and MgCl2 results plotted against the square root of the concentration, rather than the square root of the ionic strength. To correct for slightly different pH values between the measurements with different salts a small offset was added to normalize the data at zero salt. (B) Fit of the linear part of the ionic strength dependence of log k3 with the second term of Eq. 5. ▪, KCl; •, MgCl2; ▴, MgSO4; ★, NaBr. The data of all four salts were used in a common fit. ▵, ○, MgSO4 and MgCl2 results plotted against the square root of the concentration, rather than the square root of the ionic strength.
For the divalent salts MgCl2 and MgSO4 the same ionic strength is reached at three and four times lower salt concentration as for KCl. Plotted as a function of 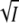 the results should fall on the same universal curves represented by Eqs. 5 and 8 and plotted in Fig. 3, A and C. We expect that at low ionic strength, when the ion-specific C-term in Eqs. 5 and 8 can be neglected, the experimental effects are caused exclusively by unspecific Debye-Hückel screening of electrostatic interactions. Transient absorption experiments in the presence of various concentrations of MgCl2 and MgSO4 were carried out at pH 7.1 (data not shown) and analyzed as described above. The low ionic strength data for the I2/I2′ equilibrium and k3 are plotted in Fig. 4, A and B, as a function of
the results should fall on the same universal curves represented by Eqs. 5 and 8 and plotted in Fig. 3, A and C. We expect that at low ionic strength, when the ion-specific C-term in Eqs. 5 and 8 can be neglected, the experimental effects are caused exclusively by unspecific Debye-Hückel screening of electrostatic interactions. Transient absorption experiments in the presence of various concentrations of MgCl2 and MgSO4 were carried out at pH 7.1 (data not shown) and analyzed as described above. The low ionic strength data for the I2/I2′ equilibrium and k3 are plotted in Fig. 4, A and B, as a function of 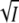 (solid circle, MgCl2; solid triangle, MgSO4) and of the square root of the MgCl2 (open circle) and MgSO4 (open triangle) concentrations. The data only fall on a straight line and agree with the results for the monovalent salts KCl (solid square) and NaBr (star) if plotted against
(solid circle, MgCl2; solid triangle, MgSO4) and of the square root of the MgCl2 (open circle) and MgSO4 (open triangle) concentrations. The data only fall on a straight line and agree with the results for the monovalent salts KCl (solid square) and NaBr (star) if plotted against 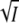 (Fig. 4, A and B, solid circle and solid triangle).
(Fig. 4, A and B, solid circle and solid triangle).
Effect of ionic strength on the amplitudes of the fluorescence from W119 in the I2 and I2′ intermediates of wild-type
The fluorescence lifetime of the unique tryptophan W119 has widely different values of 0.18, 0.82, and 0.04 ns, respectively, for the P, I2 and I2′ intermediates (5). This is due to the different extents of radiationless energy transfer between W119 and the chromophore in these states (5), which are a consequence of the different orientations of the chromophore transition dipole moments in these intermediates. Using background illumination from an LED emitting at 470 nm a photostationary mixture of P, I2, and I2′ may be accumulated. In Otto et al. (5) the contributions of these three species to the fluorescence decay curves of the mixture were measured at various pH values and it was concluded that I2 and I2′ are in a pH-dependent equilibrium with pKa ∼ 6.3. Here we performed similar experiments at fixed pH but at various salt concentrations to test whether the I2/I2′ equilibrium depends on ionic strength. Fig. 5 A shows the intensity decay curves in the dark and in the presence of background illumination at pH 8 and 1.6 M KCl. The main differences are in the early and 1–2 ns parts of the decay curve where the formation of I2′ and I2 leads to an increase in amplitude of the fastest and intermediate decay components. The slowest component with the lifetime of ∼4.8 ns is due to a small contamination with apo-PYP (5). The intensities are plotted logarithmically in Fig. 5 A, which makes it difficult to recognize the magnitude of the difference in the fastest part of the decay curve. To magnify the effect and to focus only on the changes, the light minus dark differences are plotted in Fig. 5 B on a linear intensity scale. The measurements were performed at nine different KCl concentrations. For clarity only four traces are plotted in Fig. 5 B. The two decaying components in this light-dark difference, the fastest and the slowest, are due to I2′ and I2, respectively. The rising component with the intermediate lifetime is due to P. The data in Fig. 5 B show that the amplitudes of the contributions of I2′ and I2 are salt dependent. Normalizing the I2 and I2′ amplitudes to the fraction cycling, the salt dependence of log K is plotted against 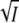 in Fig. 3 A (open square) to allow a comparison with the corresponding results from transient absorption spectroscopy (solid square). The salt dependence is qualitatively the same from both methods: at low ionic strength the I2/I2′ equilibrium is shifted toward I2′, whereas at high ionic strength a reversal occurs. The fluorescence data thus provide strong and independent support for the results obtained from time-resolved transient absorption spectroscopy. We verified that the KCl concentration had no effect on the fluorescence decay of free tryptophan at pH 8 (measurements at 0 and 1.5 M KCl).
in Fig. 3 A (open square) to allow a comparison with the corresponding results from transient absorption spectroscopy (solid square). The salt dependence is qualitatively the same from both methods: at low ionic strength the I2/I2′ equilibrium is shifted toward I2′, whereas at high ionic strength a reversal occurs. The fluorescence data thus provide strong and independent support for the results obtained from time-resolved transient absorption spectroscopy. We verified that the KCl concentration had no effect on the fluorescence decay of free tryptophan at pH 8 (measurements at 0 and 1.5 M KCl).
FIGURE 5.

(A) Fluorescence intensity decay curves of W119 of wild-type PYP in P (dark) and in a photostationary mixture of P, I2, and I2′ (light) accumulated by background illumination from an LED emitting at 470 nm. Excitation at 298 nm. Conditions, pH 8, 1.6 M KCl. (B) Light minus dark difference intensity decay at various concentrations of KCl as indicated. Note the linear intensity scale.
Effect of ionic strength on the photocycle kinetics and intermediate populations in the mutant K110A
The absorption spectrum of the mutant K110A is within experimental error identical to that of wild-type (data not shown). The salt dependence of the photocycle kinetics at pH 8 was investigated at the four wavelengths 340, 390, 450, and 490 nm, and 11 KCl concentrations between 0 and 1 M. For clarity data at only four salt concentrations are shown in Fig. 6 A. The salt dependence of the photocycle time traces at these wavelengths was much weaker than in wild-type (compare Fig. 6 A with Fig. 2 A). Using the wild-type spectra of I1, I2, and I2′ (7) these transient absorption data were transformed into the time courses of these intermediates as described in Materials and Methods. The wild-type spectra were used here, since the corresponding spectra for the mutant did not differ significantly (data not shown). The results are shown in Fig. 6 B, which should be compared with the corresponding results for wild-type in Fig. 2 B. Three conclusions can be drawn from this comparison. First, the salt effect on the I2/I2′ equilibrium is much smaller in K110A than in wild-type. This is not due to a lack of I2 at this pH. The time course of I2 of Fig. 6 B shows that a considerable amount of this intermediate remains at pH 8. Second, the ionic strength effect on kapp is also much reduced with respect to wild-type below 600 mM KCl. At higher salt the reversal effect remains. Third, the cycle is considerably slower than in wild-type. For a quantitative comparison log K is plotted against 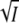 in Fig. 3 A for wild-type (solid square) and K110A (solid circle). Up to ∼600 mM KCl the equilibrium concentrations of I2 and I2′ in K110A barely change on the addition of KCl. The results for kapp are compared with those for wild-type in Fig. 7 A. Several differences are notable. The most important is that the salt dependence of kapp in K110A is much weaker than in wild-type, consistent with the smaller effect on the I2/I2′ equilibrium population (Fig. 3 A). As a quantitative measure of the salt effect on the recovery rate, we may define the change in value of kapp between low salt and the minimum in Fig. 7 A. In units of s−1 we obtain 3.9 for wild-type and 0.2 for K110A. The second difference is that kapp is about six times smaller in K110A than in wild-type below 50 mM KCl. The fact that kapp is smaller in K110A is to be expected since the attractive electrostatic interaction between the charged groups of K110 and E12 that recombine in the recovery to P is absent in the mutant. One might also predict that complete screening of the charges of K110 and E12 in wild-type would reduce kapp to the value observed for the K110A mutant. Fig. 7 A shows that this is not quite the case, at least in part because the reversal above 600 mM KCl masks the effect.
in Fig. 3 A for wild-type (solid square) and K110A (solid circle). Up to ∼600 mM KCl the equilibrium concentrations of I2 and I2′ in K110A barely change on the addition of KCl. The results for kapp are compared with those for wild-type in Fig. 7 A. Several differences are notable. The most important is that the salt dependence of kapp in K110A is much weaker than in wild-type, consistent with the smaller effect on the I2/I2′ equilibrium population (Fig. 3 A). As a quantitative measure of the salt effect on the recovery rate, we may define the change in value of kapp between low salt and the minimum in Fig. 7 A. In units of s−1 we obtain 3.9 for wild-type and 0.2 for K110A. The second difference is that kapp is about six times smaller in K110A than in wild-type below 50 mM KCl. The fact that kapp is smaller in K110A is to be expected since the attractive electrostatic interaction between the charged groups of K110 and E12 that recombine in the recovery to P is absent in the mutant. One might also predict that complete screening of the charges of K110 and E12 in wild-type would reduce kapp to the value observed for the K110A mutant. Fig. 7 A shows that this is not quite the case, at least in part because the reversal above 600 mM KCl masks the effect.
FIGURE 6.

(A) Time traces of the transient absorbance changes at 340, 390, 450, and 490 nm of the mutant K110A at pH 8 and the four selected KCl concentrations of 0 (solid line), 20 (dashed line), 200 (dotted line), and 1000 (dash-dotted line) mM. (B) Time courses of the I1, I2, I2′, and P populations derived from the data of panel A at the corresponding salt concentrations. P represents the depletion and recovery of the ground state.
FIGURE 7.

(A) Dependence of the experimental recovery rate kapp on the ionic strength (KCl) for wild-type (▪, pH 7.1), E12A (○, pH 8), and K110A (▵, pH 8). (B) pH dependence of the equilibrium concentrations of I2 (▪) and I2′ (□) in the mutant K110A. The solid curves are simultaneous fits with the Henderson-Hasselbalch equation with pKa = 5.8 and n = 1.0. Conditions, 20 mM Tris, 50 mM KCl.
The I2/I2′ equilibrium and recovery rate in wild-type are also strongly pH dependent with an apparent pKa of ∼6.3 (5,7,23). We measured the pH dependence of kapp and the I2/I2′ equilibrium in the K110A mutant at 50 mM KCl and obtained a lower pKa of ∼5.8 (Fig. 7 B). At the same pH the equilibrium in K110A is shifted further in the direction of I2′ than in wild-type, as may be expected since the interdomain interaction is weaker. The titration results show that at pH 8 a considerable amount of the I2 intermediate remains, in agreement with the time courses of Fig. 6 B.
Effect of ionic strength on the W119 fluorescence of I2′ in K110A
Further evidence for a strong reduction in the salt effect in this mutant was obtained from complementary fluorescence measurements. Fig. 8 A shows that at pH 7 in the presence of 1.5 M KCl and of background illumination, a substantial amount of both I2 and I2′ accumulates. The light minus dark intensity difference is presented in panel B on a linear scale at a number of salt concentrations. Between 0 and 0.65 M KCl the amplitudes of the I2 and I2′ contributions barely change. In spite of the presence of a considerable amount of I2 (component with slowest time constant in Fig. 8 B), salt did not shift the equilibrium in the direction of I2′. At 1.5 M KCl the amount of I2′ is reduced and that of I2 is increased (Fig. 8 B). This is similar to the high salt reversal observed in wild-type, which apparently also occurs in the absence of the salt bridge. From these data the relative amount of I2 and I2′ was calculated. The results for log K are plotted in Fig. 3 A (open circle) together with the corresponding results from flash spectroscopy (solid circle). There is good agreement between the results from these two different methods. Note that these fluorescence data for the K110A mutant were collected at pH 7, the same pH at which the wild-type flash data of Fig. 3 A were measured.
FIGURE 8.

(A) Fluorescence intensity decay curves of W119 in the mutant K110A in the dark and in the presence of background illumination (light). Excitation at 298 nm. Conditions, pH 7, 1.5 M KCl. (B) Light minus dark difference intensity decays at various KCl concentrations as indicated.
Effect of ionic strength on the photocycle kinetics of the mutant E12A
This mutation did not affect the wavelength of maximal absorbance of the chromophore. The recovery is slowed down with respect to wild-type by a factor of 2.5 at pH 8 and low salt (data not shown). Below ∼500 mM KCl the salt dependence of kapp is weaker than in wild-type as shown by the data of Fig. 7 A. Using the same measure for the salt effect as defined above for K110A, we obtain from Fig. 7 A for E12A a value of 1.2, versus 3.9 for wild-type and 0.2 for K110A. At low ionic strength the salt dependence of the recovery rate is less than in wild-type but stronger than in K110A (Fig. 7 A). At high salt the apparent recovery rate increases. This is similar to the reversal in wild-type. The effect of salt on the I2/I2′ equilibrium was measured as described by flash photolysis and fluorescence. The results are presented in Fig. 3 B, together with a fit according to Eq. 8. The data from both methods agree and show that the salt effect is similar though somewhat weaker than in wild-type. This is in line with the significant salt effect on kapp.
Effect of chaotropic anions
To gain a better understanding of the high salt reversal on the I2/I2′ equilibrium, we tested the effect of the extreme chaotropes KSCN and LiClO4. In Fig. 9 A the effect of potassium thiocyanate is compared with that of KCl. At low ionic strength thiocyanate behaves like other anions, i.e., it shifts the equilibrium toward I2′. At high salt, however, no reversal occurs and the equilibrium is shifted even further toward I2′. This in accordance with the corresponding observations on kapp, shown in Fig. 9 B, which likewise show no reversal at high ionic strength. Interestingly the slope of this salt dependence of kapp is much larger than for KCl. Similar results were obtained for 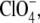 another strongly chaotropic anion (data not shown).
another strongly chaotropic anion (data not shown).
FIGURE 9.

(A) Plot of log K against  for KSCN (▪) obtained from transient absorbance data at pH 7. The fit for KCl from Fig. 3 A is shown for comparison. (B) Plot of log kapp against
for KSCN (▪) obtained from transient absorbance data at pH 7. The fit for KCl from Fig. 3 A is shown for comparison. (B) Plot of log kapp against 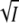 for KSCN (▪). Experimental conditions as in panel A. The fit for KCl from Fig. 3 B is shown for comparison.
for KSCN (▪). Experimental conditions as in panel A. The fit for KCl from Fig. 3 B is shown for comparison.
DISCUSSION
In the formation of the signaling state I2′ of PYP a major structural change occurs that has been characterized as partial unfolding and that involves altering the interaction between the central β-sheet and the N-terminal cap. Indirect structural evidence suggests that these two domains detach in the I2 to I2′ transition (25). Previously, we showed that salt shifts the equilibria between I1, I2, and I2′ in the mutant Y98Q toward I2/I2′ and decreases the recovery rate (18). These effects at low salt were attributed to salt binding to the salt bridge K110/E12 in the I2 to I2′ transition (18) with a binding constant of 115 mM. This interpretation of the data was tentative, since in the absence of accurate absorption spectra for I2 and I2′ it was not possible to measure the salt effect on the I2/I2′ equilibrium itself and since there was no direct evidence for such a role of the K110/E12 salt bridge. The only evidence was that the amplitude spectra at low and high salt indicated that the I2 to I2′ transition was blocked in Y98Q at low salt (18). For wild-type, the existence of a salt effect on the I1/I2/I2′ equilibria was demonstrated in Borucki et al. (18), but no systematic data were collected and in the absence of I2 and I2′ spectra, the effect on the I2/I2′ equilibrium could not be investigated. Here we show for the first time directly how salt affects the I2/I2′ equilibrium and use the K110A and E12A mutants to elucidate the role of the K110/E12 salt bridge. The reversal at high salt was attributed to the salting out property of KCl, which stabilizes the compact and folded I2 structure (18).
Based on the x-ray structure, the central β-sheet and the N-terminal cap interact by Coulomb, van der Waals, and hydrophobic interactions. Specific interactions involve the water-mediated hydrogen bonds between H108 and G7 as well as between W119 and E12, and the weak CH/π hydrogen bond between K123 and F6 (57). In addition there is the salt linkage between K110 and E12 (see Fig. 1). Residues E12, H108, K110, and W119 are highly conserved suggesting that they may play a role in the activation of PYP. The electrostatic interaction between K110 and E12 is just one of the contributions to the total interaction energy between these domains, but the short distance of 2.7 Å between the ɛ amino nitrogen of K110 and the carboxyl oxygen of E12 implies that it may make a substantial contribution. Screening of the charges of K110 and E12 by counterions or substitution of these charged residues by alanine may thus facilitate the conformational transition to the signaling state I2′ as well as slowing the recovery to P, in which this ion pair has to be reformed.
We tested these hypotheses by examining the effect of ionic strength on the I2/I2′ equilibrium as well as on the rate of recovery kapp in wild-type and in the mutants K110A and E12A. Since the spectra of the I2 and I2′ intermediates are now known (5–7), we can derive the time courses of their populations from the experimental ΔA (λ, t) traces at any ionic strength. Moreover, using the fact that the fluorescence lifetime of W119 has very different values in the I2 and I2′ intermediates, the relative amount of these species may alternatively be obtained by measuring the time-resolved fluorescence decay curves in the presence of background illumination, as described (5).
The results from these two methods were in good agreement. Up to ∼600 mM KCl the I2/I2′ equilibrium in wild-type is shifted in the direction of I2′, and at higher ionic strength a reversal back to I2 sets in. The dependence of the recovery rate on salt reflects the dependence of the I2/I2′ equilibrium on ionic strength: up to 600 mM a progressive slowing of the recovery, followed by acceleration above 600 mM. The experimental dependence of log K and log k3 on 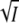 are described by two parabolas that are to a good approximation mirror images (see Fig. 3, A and C), as predicted by Eqs. 8 and 5. The good agreement between experiments and theory is unlikely to be fortuitous. Equations 5 and 8 are based on standard concepts from physical chemistry involving counterion screening of the charged groups at low salt and water binding and dehydration at high salt.
are described by two parabolas that are to a good approximation mirror images (see Fig. 3, A and C), as predicted by Eqs. 8 and 5. The good agreement between experiments and theory is unlikely to be fortuitous. Equations 5 and 8 are based on standard concepts from physical chemistry involving counterion screening of the charged groups at low salt and water binding and dehydration at high salt.
The slopes of the plots of log K and log k3 against 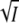 at low ionic strength (Fig. 4, A and B) allowed us to determine the product of the charges that interact. From these two sets of independent measurements we obtained values of −1.5 ± 0.2 and −1.1 ± 0.1, which are in good agreement considering the errors. The average value is close to −1, the value expected for an ion pair like K110/E12. This is a crucial result. It shows that the charges that interact and are screened are of opposite sign and most likely +1 and −1. We note that the coefficient A in Eqs. 5 and 8 is not an adjustable parameter. The fact that the charge product zA zB has this value strongly suggests that the model is applicable. The charge products from these two independent sets of measurements are in agreement thanks to the experimental fact that log k3 decreases and log K increases with ionic strength. In other words the opposite slopes in Fig. 4, A and B, are essential. The internal consistency between the two sets of experimental data and Eqs. 5 and 8 is a further argument that the model and its underlying assumptions are correct.
at low ionic strength (Fig. 4, A and B) allowed us to determine the product of the charges that interact. From these two sets of independent measurements we obtained values of −1.5 ± 0.2 and −1.1 ± 0.1, which are in good agreement considering the errors. The average value is close to −1, the value expected for an ion pair like K110/E12. This is a crucial result. It shows that the charges that interact and are screened are of opposite sign and most likely +1 and −1. We note that the coefficient A in Eqs. 5 and 8 is not an adjustable parameter. The fact that the charge product zA zB has this value strongly suggests that the model is applicable. The charge products from these two independent sets of measurements are in agreement thanks to the experimental fact that log k3 decreases and log K increases with ionic strength. In other words the opposite slopes in Fig. 4, A and B, are essential. The internal consistency between the two sets of experimental data and Eqs. 5 and 8 is a further argument that the model and its underlying assumptions are correct.
To test whether the observed salt effects depend only on the ionic strength and not on the ion species, experiments were carried out with the 2:1 electrolyte MgCl2 and the 2:2 electrolyte MgSO4. As shown in Fig. 4, A and B, when plotted against 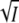 there is good agreement between the results for the monovalent salts (KCl and NaBr) and for MgCl2 and MgSO4. However, the agreement is lost, when the results are plotted against the square root of the salt concentration (Fig. 4, A and B). These observations lend further support for the idea that the effect at low ionic strength is due to unspecific counterion screening and does not depend on the cation or anion charge or species.
there is good agreement between the results for the monovalent salts (KCl and NaBr) and for MgCl2 and MgSO4. However, the agreement is lost, when the results are plotted against the square root of the salt concentration (Fig. 4, A and B). These observations lend further support for the idea that the effect at low ionic strength is due to unspecific counterion screening and does not depend on the cation or anion charge or species.
In the mutant K110A, the dependence of K and kapp on the ionic strength is virtually absent below 600 mM KCl (Figs. 3 A and 7 A). Moreover, kapp is smaller by a factor of six than in wild-type. These results strongly suggest that K110 is one of the residues that contribute to the salt bridge. PYP has many other charged groups besides K110 and E12, thus, it is not to be expected that removal of the K110/E12 salt bridge will lead to the complete disappearance of the ionic strength dependence. A residual effect is expected to remain. For the protein Fyn SH3, for example, it was recently shown that log k for folding is linear in 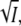 but with a positive slope (41). In this case no salt bridge was involved, but the sign was attributed to a cluster of negative surface charges (41). At high salt the reversal effect is still present in the mutant (Fig. 8 B).
but with a positive slope (41). In this case no salt bridge was involved, but the sign was attributed to a cluster of negative surface charges (41). At high salt the reversal effect is still present in the mutant (Fig. 8 B).
For E12A a significant salt dependence of K and kapp was observed, which is similar to that of wild-type. Since the side chain of K110 also makes a hydrogen bond to the backbone carbonyl of E9 (see Fig. 1 A), replacement of K110 by alanine eliminates interactions of K110 with both E9 and E12. When E12 is replaced by alanine, however, the salt bridge is lost but the hydrogen bond between K110 and E9 would be expected to remain. More importantly the ɛ-amino group of K110 can now form a novel salt bridge with the carboxyl group of E9 (see Fig. 1 A), as we showed by energy minimization and model building. This requires some movement of the E9 side chain, which can occur without a steric clash. We attribute the observed salt dependence of the I2/I2′ equilibrium and recovery rate in E12A to the presence of this new salt bridge.
Our result for the salt dependence of the recovery rate of the mutant K110A (Fig. 7 A) is consistent with those reported for the truncated PYP's T6 and T15 in which the first six or 15 N-terminal residues were removed by proteolytic digestion (17). For T6 (with the K110/E12 salt linkage presumably still intact) a minimum in the salt dependence of the recovery rate around 600 mM NaCl was observed as in wild-type. For T15 however (no salt bridge) the decrease of the recovery rate at low salt was absent, but the high salt reversal was still present. These T15 results are similar to our results for the K110A and E12A mutants reported in Fig. 7 A, which clearly indicate the reversal at 1 M KCl.
Two phases could be distinguished in the salt dependence for wild-type, the low salt phase due to screening and the reversal phase at high salt due to water binding to the salt. Eqs. 5 and 8 were based on the effects these phenomena have on the activity of the salt bridge K110A/E12A and thus appropriate for wild-type only. In the K110A and E12A mutants no effect would be expected to remain. The low salt screening effects on both the equilibrium and the recovery rates are indeed very much reduced as expected (Figs. 3 A and 7 A). A significant part of the high salt reversal phase remained, however, in the K110A and E12A mutants (Figs. 3, A and B, and 7 A) as in the T15 mutant (17). At high concentrations salt will affect the water structure and lead to salting out, whether the K110/E12 salt bridge is present or not. Salt increases the surface tension of water and thereby increases the free energy of cavity formation (42). In this way salt leads to the salting out of nonpolar groups (42). Since the I2 to I2′ conformational transition is associated with an expansion (25) and exposure of a hydrophobic surface patch (26,19), high salt is expected to favor the compact and folded structure of the I2 intermediate. Salting out will thus also shift the I2/I2′ equilibrium back in the direction of I2. We attribute the reversal effects in the mutants at high salt to these salting out effects. The third terms in Eqs. 5 and 8 for wild-type thus contain also salting out contributions, for which a linear dependence on the salt concentration is expected (41,42).
At high salt specific anion effects are expected to occur. Anions are ordered according to the strength of these effects in the Hofmeister series (42). Our results with kosmotropic anions like Cl− and 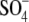 showed that these favor the compact folded I2 intermediate. For the strongly chaotropic anions SCN− and
showed that these favor the compact folded I2 intermediate. For the strongly chaotropic anions SCN− and 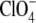 we found the opposite effect (Fig. 9 A): these anions shifted the I2/I2′ equilibrium toward the unfolded form I2′, which has a larger radius of gyration. Extreme chaotropes like SCN− also affect the initial dark state of PYP, shifting the ground state equilibrium in wild-type and the mutant Y42F toward a population with a protonated chromophore, which is less stabile and resembles the photocycle intermediate I2′ (43–45).
we found the opposite effect (Fig. 9 A): these anions shifted the I2/I2′ equilibrium toward the unfolded form I2′, which has a larger radius of gyration. Extreme chaotropes like SCN− also affect the initial dark state of PYP, shifting the ground state equilibrium in wild-type and the mutant Y42F toward a population with a protonated chromophore, which is less stabile and resembles the photocycle intermediate I2′ (43–45).
Taken together our results strongly suggest that the salt bridge K110/E12 plays a key role in keeping PYP in the inactive form in the dark and that this salt bridge needs to be broken on light-induced activation. The I2/I2′ equilibrium may be compared to the MI/MII equilibrium in the photoreceptor rhodopsin, where high salt also favors the signaling state, MII. It has been proposed that an ionic lock involving the conserved motif ERY of helix 3 and E247 of helix 6 holds these helices together in the dark (46). In MII, these helices move apart (47,48), and this conformational change is a universal feature of G-protein coupled receptors (49), which share a common mechanism of activation (50). In the case of PYP our data strongly suggest that the N-terminal domain detaches from the β-scaffold of the PAS core in I2′, but direct structural evidence that these domains move apart is still lacking.
A change in domain interactions also plays a significant role in the activation of another PAS domain protein, the LOV2 domain of phototropin. High resolution solution structures were determined for the dark and the activated state (51,52). In this photoreceptor a light-induced distortion of the central β-sheet leads to dissociation of the C-terminal Jα helix from the PAS core and activation of the kinase activity (51,52). It seems that in PYP an analogous change in domain-domain interaction occurs in the formation of the signaling state but involving the detachment of the N-terminal cap from the central β-sheet. However, no salt bridge contributes to the domain-domain interaction in LOV2 (52).
Few experimental results are available on the effect of ionic strength on the rate of protein folding. However, with hen lysozyme the slowest folding rate constant (∼1 s−1) slowed with increasing NaCl concentration and displayed the same linear dependence of log k on 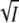 as observed here with PYP, i.e., with a negative slope (39). It was concluded that this slow phase of the folding is associated with the formation of a specific attractive ionic interaction (39). T4 lysozyme has an interdomain interaction that involves hinge-bending and the disruption of an interdomain salt bridge (40). In the open conformation the stabilizing salt bridge between E22 and R137, which links the two domains in the closed conformation, is broken and the distance between the charged groups increases to >17 Å (40). It seems likely that, as observed here for PYP, the formation of the closed form and the salt bridge is slowed by counterion screening of the Coulomb attraction.
as observed here with PYP, i.e., with a negative slope (39). It was concluded that this slow phase of the folding is associated with the formation of a specific attractive ionic interaction (39). T4 lysozyme has an interdomain interaction that involves hinge-bending and the disruption of an interdomain salt bridge (40). In the open conformation the stabilizing salt bridge between E22 and R137, which links the two domains in the closed conformation, is broken and the distance between the charged groups increases to >17 Å (40). It seems likely that, as observed here for PYP, the formation of the closed form and the salt bridge is slowed by counterion screening of the Coulomb attraction.
It is of interest to compare our observations for a salt bridge between two protein domains with those for the related problem of the electrostatic interaction between two proteins. With oppositely charged interacting domains the association rate constant kas and the equilibrium constant K decreased with ionic strength for the thrombin-hirudin and the barnase-barstar complexes (53–55) in the same way as observed here for PYP below 600 mM KCl. Changing the charge product of the two proteins by mutagenesis of charged residues in the interface region led to qualitative agreement with models with linear terms in  as in Eqs. 5 and 8 (53–55). This is similar to our observation with the K110A mutant of PYP. We note that in the case of the thrombin-hirudin complex plots of log K and log kas against
as in Eqs. 5 and 8 (53–55). This is similar to our observation with the K110A mutant of PYP. We note that in the case of the thrombin-hirudin complex plots of log K and log kas against 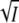 showed clear curvature (55), which in the case of PYP could be described by the term proportional to I in Eqs. 5 and 8. In the work on the barnase-barstar complex an equation was proposed for log kas which included only the linear term in
showed clear curvature (55), which in the case of PYP could be described by the term proportional to I in Eqs. 5 and 8. In the work on the barnase-barstar complex an equation was proposed for log kas which included only the linear term in 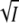 of Eq. 5 (54). For ionic strength below 100 mM the agreement was good, but curvature was observed here as well at higher salt concentration, which may well be accounted for by a term proportional to I as in Eq. 5.
of Eq. 5 (54). For ionic strength below 100 mM the agreement was good, but curvature was observed here as well at higher salt concentration, which may well be accounted for by a term proportional to I as in Eq. 5.
To our knowledge an analysis of the salt dependence of a protein conformational equilibrium (I2/I2′) and of a recovery (refolding) rate constant based on Eqs. 5 and 8 has not been reported. In protein folding the formation of interdomain salt bridges plays a significant role and the salt dependence of the stability of these salt bridges is likely of importance. The conceptual framework presented here may thus be applicable to related problems concerning the ionic strength dependence of protein structure and dynamics.
Acknowledgments
We thank Elsa Chan for making the K110A and E12A mutants.
This work was financially supported by grants from the National Institutes of Health (GM 66126 to M.A.C.) and the Deutsche Forschungsgemeinschaft (He 1382/14-1 to M.P.H. and H.O.).
Abbreviations used: PAS domain, acronym formed from the names of the first three proteins recognized as sharing this sensory domain; PYP, photoactive yellow protein.
Editor: Janos K. Lanyi.
References
- 1.Cusanovich, M. A., and T. E. Meyer. 2003. Photoactive yellow protein: a prototypic PAS domain sensory protein and development of a common signaling mechanism. Biochemistry. 42:4759–4770. [DOI] [PubMed] [Google Scholar]
- 2.Taylor, B. L., and I. B. Zhulin. 1999. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol. Mol. Biol. Rev. 63:479–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heyne, K., O. F. Mohammed, A. Usman, J. Dreya, E. T. J. Nibbering, and M. A. Cusanovich. 2005. Structural evolution of the chromophore in the primary stages of the trans/cis isomerization in photoactive yellow protein. J. Am. Chem. Soc. 127:18100–18106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoff, W. D., I. H. M. van Stokkum, H.-J. van Ramesdonk, M. E. van Brederode, A. M. Brouwer, J. C. Fitch, T. E. Meyer, R. van Grondelle, and K. J. Hellingwerf. 1994. Measurement and global analysis of the absorbance changes in the photocycle of the photoactive yellow protein from Ectothiorhodospira halophila. Biophys. J. 67:1691–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otto, H., D. Hoersch, T. E. Meyer, M. A. Cusanovich, and M. P. Heyn. 2005. Time-resolved single tryptophan fluorescence in photoactive yellow protein monitors changes in the chromophore structure during the photocycle via energy transfer. Biochemistry. 44:16804–16816. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu, N., Y. Imamoto, M. Harigai, H. Kamikubo, Y. Yamazaki, and M. Kataoka. 2006. pH-dependent equilibrium between long lived near-UV intermediates of photoactive yellow protein. J. Biol. Chem. 281:4318–4325. [DOI] [PubMed] [Google Scholar]
- 7.Borucki, B., C. P. Joshi, H. Otto, M. A. Cusanovich, and M. P. Heyn. 2006. The transient accumulation of the signaling state of photoactive yellow protein is controlled by the external pH. Biophys. J. 91:2991–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee, B.-C., P. A. Croonquist, T. R. Sosnick, and W. D. Hoff. 2001. PAS domain receptor photoactive yellow protein is converted to a molten globule state upon activation. J. Biol. Chem. 276:20821–20823. [DOI] [PubMed] [Google Scholar]
- 9.Xie, A., L. Kelemen, J. Hendriks, B. J. White, K. J. Hellingwerf, and W. D. Hoff. 2001. Formation of a new buried charge drives a large-amplitude protein quake in photoreceptor activation. Biochemistry. 40:1510–1517. [DOI] [PubMed] [Google Scholar]
- 10.Brudler, R., R. Rammelsberg, T. T. Woo, E. D. Getzoff, and K. Gerwert. 2001. Structure of the I1 early intermediate of photoactive yellow protein by FTIR spectroscopy. Nat. Struct. Biol. 8:265–270. [DOI] [PubMed] [Google Scholar]
- 11.Rubinstenn, G., G. W. Vuister, F. A. A. Mulder, P. E. Düx, R. Boelens, K. J. Hellingwerf, and R. Kaptein. 1998. Structural and dynamic changes of photoactive yellow protein during its photocycle in solution. Nat. Struct. Biol. 5:568–570. [DOI] [PubMed] [Google Scholar]
- 12.Craven, C. J., N. M. Derix, J. Hendriks, R. Boelens, K. J. Hellingwerf, and R. Kaptein. 2000. Probing the nature of the blue-shifted intermediate of photoactive yellow protein in solution by NMR: hydrogen-deuterium exchange data and pH studies. Biochemistry. 39:14392–14399. [DOI] [PubMed] [Google Scholar]
- 13.Borgstahl, G. E. O., D. R. Williams, and E. D. Getzoff. 1995. 1.4 Å structure of photoactive yellow protein, a cytosolic photoreceptor: unusual fold, active site and chromophore. Biochemistry. 34:6278–6287. [DOI] [PubMed] [Google Scholar]
- 14.Genick, U. K., G. E. Borgstahl, K. Ng, Z. Ren, C. Pradervand, P. M. Burke, V. Srajer, T. Y. Teng, W. Schildkamp, D. E. McRee, K. Moffat, and E. D. Getzoff. 1997. Structure of a protein photocycle intermediate by millisecond time-resolved crystallography. Science. 275:1471–1475. [DOI] [PubMed] [Google Scholar]
- 15.Ihee, H., S. Rajagopal, V. Šrajer, R. Pahl, S. Anderson, M. Schmidt, F. Schotte, P. A. Anfinrud, M. Wulff, and K. Moffat. 2005. Visualizing reaction pathways in photoactive yellow protein from nanoseconds to seconds. Proc. Natl. Acad. Sci. USA. 102:7145–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dux, P., G. Rubinstenn, G. W. Vuister, R. Boelens, F. A. A. Mulder, K. Hard, W. D. Hoff, A. R. Kroon, W. Crielaard, K. J. Hellingwerf, and R. Kaptein. 1998. Solution structure and backbone dynamics of the photoactive yellow protein. Biochemistry. 37:12689–12699. [DOI] [PubMed] [Google Scholar]
- 17.Harigai, M., Y. Imamoto, H. Kamikubo, Y. Yamazaki, and M. Kataoka. 2003. Role of an N-terminal loop in the secondary structural change of photoactive yellow protein. Biochemistry. 42:13893–13900. [DOI] [PubMed] [Google Scholar]
- 18.Borucki, B., J. A. Kyndt, C. P. Joshi, H. Otto, T. E. Meyer, M. A. Cusanovich, and M. P. Heyn. 2005. Effect of salt and pH on the activation of photoactive yellow protein and gateway mutants Y98Q and Y98F. Biochemistry. 44:13650–13663. [DOI] [PubMed] [Google Scholar]
- 19.Borucki, B., S. Devanathan, H. Otto, M. A. Cusanovich, G. Tollin, and M. P. Heyn. 2002. Kinetics of proton uptake and dye binding by photoactive yellow protein in wildtype and in the E46Q and E46A mutants. Biochemistry. 41:10026–10037. [DOI] [PubMed] [Google Scholar]
- 20.Hendriks, J., I. H. M. van Stokkum, and K. J. Hellingwerf. 2003. Deuterium isotope effects in the photocycle transitions of the photoactive yellow protein. Biophys. J. 84:1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borucki, B., H. Otto, C. P. Joshi, C. Gasperi, M. A. Cusanovich, S. Devanathan, G. Tollin, and M. P. Heyn. 2003. pH dependence of the photocycle kinetics of the E46Q mutant of photoactive yellow protein: protonation equilibrium between the I1 and I2 intermediates, chromophore deprotonation by hydroxyl uptake, and protonation relaxation in the dark state. Biochemistry. 42:8780–8790. [DOI] [PubMed] [Google Scholar]
- 22.Joshi, C. P., B. Borucki, H. Otto, T. E. Meyer, M. A. Cusanovich, and M. P. Heyn. 2006. Photocycle and photoreversal of photoactive yellow protein at alkaline pH: kinetics, intermediates and equilibria. Biochemistry. 45:7057–7068. [DOI] [PubMed] [Google Scholar]
- 23.Imamoto, Y., M. Harigai, and M. Kataoka. 2004. Direct observation of the pH-dependent equilibrium between L-like and M intermediates of photoactive yellow protein. FEBS Lett. 577:75–80. [DOI] [PubMed] [Google Scholar]
- 24.Joshi, C. P., B. Borucki, H. Otto, T. E. Meyer, M. A. Cusanovich, and M. P. Heyn. 2005. Photoreversal kinetics of the I1 and I2 intermediates in the photocycle of photoactive yellow protein by double flash experiments with variable time delay. Biochemistry. 44:656–665. [DOI] [PubMed] [Google Scholar]
- 25.Imamoto, Y., H. Kamikubo, M. Harigai, N. Shimizu, and M. Kataoka. 2002. Light-induced global conformational change of photoactive yellow protein in solution. Biochemistry. 41:13595–13601. [DOI] [PubMed] [Google Scholar]
- 26.Meyer, T. E., G. Tollin, J. H. Hazzard, and M. A. Cusanovich. 1989. Photoactive yellow protein from the purple phototrophic bacterium Ectothiorhodospira halophila. Quantum yield of photobleaching and effects of temperature, alcohols, glycerol and sucrose on kinetics of photobleaching and recovery. Biophys. J. 56:559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Brederode, M., W. D. Hoff, I. H. M. Van Stokkum, M.-L. Groot, and K. J. Hellingwerf. 1996. Protein folding thermodynamics applied to the photocycle of the photoactive yellow protein. Biophys. J. 71:365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, B.-C., P. A. Croonquist, and W. D. Hoff. 2001. Mimic of photocycle by a protein folding reaction in photoactive yellow protein. J. Biol. Chem. 276:44481–44487. [DOI] [PubMed] [Google Scholar]
- 29.Jiang, Z. Y., L. R. Swem, B. G. Rushing, S. Devanathan, G. Tollin, and C. E. Bauer. 1999. Bacterial photoreceptor with similarity to photoactive yellow protein and plant phytochromes. Science. 285:406–409. [DOI] [PubMed] [Google Scholar]
- 30.Kyndt, J. A., J. C. Fitch, T. E. Meyer, and M. A. Cusanovich. 2005. Thermochromatium tepidum photoactive yellow protein/bacteriophytochrome/diguanylate cyclase Ppd.: characterization of the PYP domain. Biochemistry. 44:4755–4764. [DOI] [PubMed] [Google Scholar]
- 31.Crosson, S., S. Rajagopal, and K. Moffat. 2003. The LOV domain family: photoresponsive signaling modules coupled to diverse output domains. Biochemistry. 42:2–10. [DOI] [PubMed] [Google Scholar]
- 32.Losi, A., E. Ghiraldelli, S. Jansen, and W. Gärtner. 2005. Mutational effects on protein structural changes and interdomain interactions in the blue-light sensing LOV protein YtvA. Photochem. Photobiol. 81:1145–1152. [DOI] [PubMed] [Google Scholar]
- 33.Kyndt, J. A., F. Vanrobaeys, J. C. Fitch, B. V. Devreese, T. E. Meyer, M. A. Cusanovich, and J. J. Van Beeumen. 2003. Heterologous production of Halorhodospira halophila holo-photoactive yellow protein through tandem expression of the postulated biosynthetic genes. Biochemistry. 42:965–970. [DOI] [PubMed] [Google Scholar]
- 34.Kyndt, J. A., J. K. Hurley, B. Devreese, T. E. Meyer, M. A. Cusanovich, G. Tollin, and J. J. Van Beeumen. 2004. Rhodobacter capsulatus photoactive yellow protein: genetic context, spectral and kinetics characterization, and mutagenesis. Biochemistry. 43:1809–1820. [DOI] [PubMed] [Google Scholar]
- 35.Borucki, B., H. Otto, and M. P. Heyn. 1999. Reorientation of the retinylidene chromophore in the K, L, and M intermediates of bacteriorhodopsin from time-resolved linear dichroism: resolving kinetically and spectrally overlapping intermediates of chromoproteins. J. Phys. Chem. B. 103:6371–6383. [Google Scholar]
- 36.Atkins, P. W. 1998. Physical Chemistry. Oxford University Press, Oxford, UK.
- 37.Bockris, J. O'M., and A.K.N. Reddy. 1998. Modern Electrochemistry, Vol. 1, 2nd Ed. Plenum Press, New York.
- 38.Stokes, R. H., and R. A. Robinson. 1948. Ionic hydration and activity in electrolyte solutions. J. Am. Chem. Soc. 70:1870–1878. [DOI] [PubMed] [Google Scholar]
- 39.Itzhaki, L. S., P. A. Evans, C. M. Dobson, and S. E. Radford. 1994. Tertiary interactions in the folding pathway of hen lysozyme: kinetic studies using fluorescence probes. Biochemistry. 33:5212–5220. [DOI] [PubMed] [Google Scholar]
- 40.Sinha, N., S. Kumar, and R. Nussinov. 2001. Interdomain interactions in hinge-bending transitions. Structure. 9:1165–1181. [DOI] [PubMed] [Google Scholar]
- 41.de los Rios, M. A., and K. W. Plaxco. 2005. Apparent Debye-Hückel electrostatic effects in the folding of a simple, single domain protein. Biochemistry. 44:1243–1250. [DOI] [PubMed] [Google Scholar]
- 42.Baldwin, R. L. 1996. How Hofmeister ion interactions affect protein stability. Biophys. J. 71:2056–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brudler, R., T. E. Meyer, U. K. Genick, S. Devanathan, T. T. Woo, D. P. Millar, K. Gerwert, M. A. Cusanovich, G. Tollin, and E. D. Getzoff. 2000. Coupling of hydrogen bonding to chromophore conformation and function in photoactive yellow protein. Biochemistry. 39:13478–13486. [DOI] [PubMed] [Google Scholar]
- 44.Meyer, T. E., S. Devanathan, T. Woo, E. D. Getzoff, G. Tollin, and M. A. Cusanovich. 2003. Site specific mutations provide new insight into the origin of pH effects and alternative spectral forms in the photoactive yellow protein of Halorhodospira halophila. Biochemistry. 42:3319–3325. [DOI] [PubMed] [Google Scholar]
- 45.El-Mashtoly, S. F., M. Unno, M. Kumauchi, N. Hamada, K. Fujiwara, J. Sasaki, Y. Imamoto, M. Kataoka, F. Tokunaga, and S. Yamauchi. 2004. Resonance Raman spectroscopy reveals the origin of an intermediate wavelength form in photoactive yellow protein. Biochemistry. 43:2279–2287. [DOI] [PubMed] [Google Scholar]
- 46.Vogel, R., and F. Siebert. 2002. Conformation and stability of α-helical membrane proteins. 1. Influence of salts on conformational equilibria between active and inactive states of rhodopsin. Biochemistry. 41:3529–3535. [DOI] [PubMed] [Google Scholar]
- 47.Farrens, D. L., C. Altenbach, K. Yang, W. L. Hubbell, and H. G. Khorana. 1996. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 274:768–770. [DOI] [PubMed] [Google Scholar]
- 48.Sheikh, S. P., T. A. Zvyaga, O. Lichtarge, T. P. Sakmar, and H. R. Bourne. 1996. Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature. 383:347–350. [DOI] [PubMed] [Google Scholar]
- 49.Ghadouni, P., J. J. Steenhuis, D. L. Farrens, and B. K. Kobilka. 2001. Agonist-induced conformational changes in the G-protein-coupling domain of the β2 adrenergic receptor. Proc. Natl. Acad. Sci. USA. 98:5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ballesteros, J. A., A. D. Jensen, G. Liapakis, S. G. Rasmussen, L. Shi, U. Gether, and J. A. Javitch. 2001. Activation of the β2 adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 276:27171–27177. [DOI] [PubMed] [Google Scholar]
- 51.Harper, S. M., L. C. Neil, and K. H. Gardner. 2003. Structural basis of a phototropin light switch. Science. 301:1541–1544. [DOI] [PubMed] [Google Scholar]
- 52.Harper, S. M., J. M. Christie, and K. H. Gardner. 2004. Disruption of the LOV-Jα helix interaction activates phototropin kinase activity. Biochemistry. 43:16184–16192. [DOI] [PubMed] [Google Scholar]
- 53.Schreiber, G., and A. R. Fersht. 1996. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 3:427–431. [DOI] [PubMed] [Google Scholar]
- 54.Vijayakumar, M., K.-Y. Wong, G. Schreiber, A. R. Fersht, A. Szabo, and H.-X. Zhou. 1998. Electrostatic enhancement of diffusion-controlled protein-protein association: comparison of theory and experiment on barnase and barstar. J. Mol. Biol. 278:1015–1024. [DOI] [PubMed] [Google Scholar]
- 55.Stone, S. R., S. Dennis, and J. Hofsteenge. 1989. Quantitative evaluation of the contribution of ionic interactions to the formation of the thrombin-hirudin complex. Biochemistry. 28:6857–6863. [DOI] [PubMed] [Google Scholar]
- 56.Getzoff, E. D., K. N. Gutwin, and U. K. Genick. 2003. Anticipatory active-site motions and chromophore distortion prime photoreceptor PYP for light activation. Nat. Struct. Biol. 10:663–668. [DOI] [PubMed] [Google Scholar]
- 57.Harigai, M., M. Kataoka, and Y. Imamoto. 2006. A single CH/π weak hydrogen bond governs stability and the photocycle of the photoactive yellow protein. J. Am. Chem. Soc. 128:10646–10647. [DOI] [PubMed] [Google Scholar]