

Abstract
This study demonstrates that the β-chemokines macrophage inflammatory proteins 1α and 1β (MIP-1α and MIP-1β) and, RANTES (regulated on activation, normally T-cell expressed and secreted) inhibit human immunodeficiency virus (HIV) replication in anti-CD3 or recall antigen-stimulated peripheral blood mononuclear cells (PBMCs) of asymptomatic HIV-infected subjects. Significant levels of β-chemokines were produced by both CD4+ and CD8+ PBMC subsets from HIV-infected individuals. Neutralization of endogenous MIP-1α, MIP-1β, and RANTES did not rescue HIV replication in cultures to which greater than 10% CD8+ T cells had been added, indicating that the HIV suppressor activity of CD8+ T cells cannot be explained entirely by the β-chemokines. However, significant enhancement of viral replication was observed upon neutralization of endogenous β-chemokines in CD8-depleted or CD4+ PBMCs from most donors, particularly in cultures with low inducible levels of HIV production. In contrast, certain endogenous proinflammatory cytokines induced HIV replication in these same cells. These data suggest that the levels of HIV replication in CD4+ PBMC reflect the balance of the opposing effects of endogenous suppressive factors, such as the β-chemokines, and HIV-inducing cytokines, such as tumor necrosis factor α and interleukin 1β.
Keywords: proinflammatory, regulation
The regulation of human immunodeficiency virus (HIV) replication by the network of endogenous cytokines is enormously complex (reviewed in ref. 1). Certain cytokines, such as interferon α (2) and interleukin (IL)-10 (3, 4), primarily down-regulate virus replication, whereas others, particularly the proinflammatory cytokines tumor necrosis factor (TNF)-α (5, 6, 7) and IL-1β (7, 8, 9, 10), have been found to enhance HIV production in vitro. Cytokines may act synergistically (9, 11) or antagonistically (3, 10, 12) to regulate HIV expression/replication; the overall effect on HIV production being dependent upon the cytokines present in a particular microenvironment. Recent studies (13, 14, 15, 16, 17) demonstrate that the chemokines, a superfamily of chemotactic factors involved in the recruitment and activation of leukocytes during inflammation (reviewed in ref. 18), can now be included in the group of factors that regulate HIV replication and spread. These studies demonstrated that certain members of the β-chemokine family, namely macrophage inflammatory protein-1α and 1β (MIP-1α and MIP-1β), and RANTES (regulated on activation, normally T-cell expressed and secreted), suppress the replication of macrophage-tropic, but not T-cell-tropic, HIV strains in in vitro infected mitogen-activated primary T cells from uninfected donors or T-cell lines. The mechanism for this inhibition is now known to be related to the downregulation or blocking by these chemokines of the C-C chemokine receptor-5 (CCR-5), which has recently been identified as a necessary coreceptor used by macrophage tropic HIV strains for fusion with the cell membrane (15, 16, 17). However, the role that β-chemokines may play in the regulation of virus replication and spread in in vivo infected cells from HIV-infected subjects has yet to be determined.
This study demonstrates that β-chemokines exert an inhibitory effect on HIV replication in primary CD8-depleted and CD4+ peripheral blood mononuclear cells (PBMCs) from HIV-infected individuals stimulated in vitro with recall antigen or anti-CD3 antibody. CD8-depleted and CD4+ PBMCs of most asymptomatic HIV-infected subjects tested produce substantial levels of β-chemokines, which play a significant role in controlling HIV replication and spread in vitro, as determined by neutralization assays. Furthermore, the levels of HIV replication in CD4+ PBMC cultures were found to reflect a balance of the effects of endogenous HIV-suppressive factors, such as the β-chemokines, and those of endogenous HIV-inducing cytokines, such as TNF-α and IL-1. Finally, the β-chemokines cannot fully account for the CD8+ T-cell-mediated suppression of HIV replication in PBMC from HIV-infected subjects.
MATERIALS AND METHODS
Cellular Populations.
PBMCs were obtained from apheresis of 20 HIV-infected individuals (CD4+ T-cell count: range 159–885/μl; mean 495/μl) after separation over Ficoll Hypaque density gradients. PBMCs were separated into CD4-depleted or CD8-depleted subsets (>96% depleted as determined by FACS analysis) using immunomagnetic beads (Dynal, Great Neck, NY); CD4+ and CD8+ T-cell subsets (>96% pure as determined by FACS analysis) were obtained by depletion of either CD4+ or CD8+ cells from E-rosette (+) T cells, unless otherwise indicated. Monocyte/macrophages were obtained by adherence for 45 min of CD8-depleted PBMCs onto flasks followed by five vigorous washes in PBS and gentle scraping.
Effect of Exogenous β-Chemokines.
CD8-depleted PBMCs were cultured at 1.5–2 × 106 per well in 48-well plates in RPMI medium with 10% fetal calf serum (endotoxin <10 pg/ml), supplemented with 1 mM antibiotics, glutamine, and Hepes buffer. Cultures were treated with various concentrations (0.5–100 ng/ml) of β-chemokines [recombinant human (rh) MIP-1α, rhMIP-1β, rhRANTES, rhMCP-1, or rhIL-8; R & D Systems], added individually or in combination and stimulated with either anti-CD3 (mouse ascites, 1:4000 dilution) plus IL-2 (10 units/ml; Boehringer Mannheim) or with tetanus toxoid (12.5 μg/ml; Wyeth Ayerst Laboratories, Marietta, PA). Cultures were refed with β-chemokines alone (recall antigen cultures) or with the addition of IL-2 (10 units/ml) (anti-CD3-stimulated cultures) twice weekly.
Endogenous β-Chemokine Assays.
CD8-depleted PBMCs, cultured alone as described above or with various proportions of autologous CD8+ T cells, were cultured in the absence or presence of IgG isotype control mAb antibodies (R & D Systems) or neutralizing antibodies directed against β-chemokines, individually or in combination [unless otherwise indicated: polyclonal anti-MIP-1α (50 μg/ml), polyclonal anti-MIP-1β (50 μg/ml), and monoclonal anti-RANTES (10 μg/ml); R & D Systems] immediately prior to stimulation of cultures with either anti-CD3 plus IL-2 or recall antigen as described above. Cultures were refed twice weekly with antibodies and, in anti-CD3-stimulated cultures, with IL-2. CD4+ T cells were supplemented with an additional 5% monocyte/macrophages and cultured in anti-CD3 plus IL-2-stimulated conditions in the presence of either isotype control antibodies or a combination of the anti-β-chemokine antibodies (as described above), a combination of antagonists of proinflammatory cytokines [IL-1ra (200 ng/ml), sTNFR (10 μg/ml), and goat anti-IL-6 (5 μg/ml); R & D Systems] or both. Cultures were refed with antibodies and maintained as described above.
Analysis of β-Chemokine Production.
Unfractionated, CD8-depleted, CD4-depleted, and negatively or positively selected CD4+ and CD8+ T-cell subsets were cultured at 2 × 106/ml and left untreated or stimulated with phytohemagglutinin (PHA) (4 μg/ml). Culture supernatants were harvest at 6, 18, 24, 48, and 72 h and 5 and 7 days after stimulation and frozen at −80°C for later analysis of β-chemokine production by ELISA for MIP-1α, MIP-1β, and RANTES (R & D Systems).
Quantitation of HIV.
Culture supernatants were analyzed for levels of HIV either by reverse transcriptase assay, as previously described (19), or by HIV p24 ELISA (DuPont).
RESULTS
The Effect of Exogenous MIP-1α, MIP-1β, and RANTES on HIV Replication in CD8-Depleted or CD4+ PBMCs of HIV-Infected Subjects.
The effect of exogenous β-chemokines on HIV replication was assessed in CD8-depleted PBMCs from 15 HIV-infected subjects (CD4+ T cells 154–885/μl; mean = 495/μl), five of whom had been recently boosted with tetanus toxoid (TT). Various concentrations of MIP-1α, MIP-1β, or RANTES were added with either recall antigen (TT) or anti-CD3 plus IL-2. MIP-1α, MIP-1β, and RANTES dramatically suppressed HIV replication and this effect was often obtained, particularly in TT-stimulated conditions (Fig. 1A), at chemokine concentrations (5 ng/ml) 100-fold less than those previously shown to effectively inhibit HIV replication in in vitro-infected, mitogen-stimulated PBMCs or T-cell lines (13, 17). Addition of β-chemokines (10–100 ng/ml) inhibited HIV replication induced by either recall antigen (TT) or anti-CD3 by 45–95% in the majority (12 of 15) of donor PBMCs (Fig. 1B); the β-chemokine macrophage chemotactic protein-1 and the α-chemokine IL-8 either had no effect or slightly enhanced HIV replication (data not shown). While the degree of inhibition of HIV replication exerted by a particular chemokine varied among PBMCs from different HIV-infected donors, RANTES consistently produced the most dramatic inhibition (Fig. 1B). Of interest, β-chemokine-mediated inhibition of HIV replication frequently did not exhibit linear dose dependence, particularly in recall antigen-stimulated conditions (data not shown). β-chemokine-mediated inhibition of HIV replication was not related to suppression of CD4+ T-cell activation as determined by the induction of IL-2 production, expression of CD25 (IL-2Rα) on CD4+ T cells, and cellular proliferation in recall antigen-stimulated cultures (data not shown).
Figure 1.
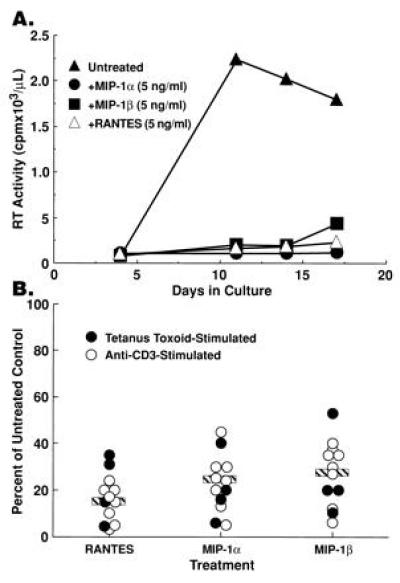
Exogenous β-chemokines inhibit HIV replication in CD8-depleted PBMCs from HIV-infected subjects. (A) Supernatant-associated reverse transcriptase activity present in cultures of CD8-depleted PBMC stimulated with tetanus toxoid in the absence or presence of 5 ng/ml of either rhMIP-1α, rhMIP-1β, or RANTES. (B) Summary of the reduction of peak levels of in vitro HIV replication upon treatment of CD8-depleted PBMCs from 12 HIV-infected individuals with MIP-1α (x̄ = 23% of control), MIP-1β (x̄ = 28% of control), or RANTES (x̄ = 16% of control) (β-chemokines used at 0.5–100 ng/ml).
Numerous PBMC Subsets Produce β-Chemokines.
To determine which PBMC subsets were the primary source of endogenous β-chemokine production, unfractionated, CD4-depleted, CD8-depleted or CD4+ and CD8+ PBMC subsets from HIV-infected subjects were assayed for the secretion of MIP-1α, MIP-1β, and RANTES under various stimulatory conditions. Unfractionated, CD8-depleted and CD4-depleted PBMC produced comparable levels of β-chemokines under most conditions (data not shown). Significant upregulation of β-chemokine production in response to PHA was observed in both CD4+ and CD8+ T cells (Table 1). Of interest, β-chemokine production by negatively selected CD4+ T cells was equal to or often greater than levels produced by parallel cultures of autologous CD8+ T cells (Table 1). Levels of β-chemokines produced in PHA-stimulated conditions by either CD4+ or CD8+ PBMC subsets of asymptomatic HIV-infected subjects did not differ significantly from levels produced by those of HIV-uninfected donors (data not shown). Recall antigen stimulation of CD8-depleted PBMC from HIV-infected donors resulted in significantly lower levels of β-chemokine production (2- to 10-fold), and the peak production of β-chemokines was delayed compared with that observed with more potent cellular activators such as PHA or anti-CD3 (data not shown).
Table 1.
Production of MIP-1α, MIP-1β, and RANTES (pg/ml) by unfractionated, CD8+, and CD4+ PBMC from three HIV-infected subjects
| Unfractionated
|
CD8+
|
CD4+
|
||||
|---|---|---|---|---|---|---|
| Unstimulated | PHA | Unstimulated | PHA | Unstimulated | PHA | |
| MIP-1α | ||||||
| 1 | 40 | 7,418 | 10 | 2,693 | 10 | 3,835 |
| 2* | 13 | 1,531 | 10 | 4,396 | 10 | 733 |
| 3 | 10 | 8,138 | 224 | 3,201 | 1729 | 6,580 |
| MIP-1β | ||||||
| 1 | 838 | 19,305 | 59 | 20,975 | 46 | 20,308 |
| 2* | 104 | 24,928 | 123 | 28,357 | 19 | 4,094 |
| 3 | 10 | 29,773 | 171 | 23,759 | 1738 | 28,557 |
| RANTES | ||||||
| 1 | 10 | 4,759 | 10 | 1,914 | 10 | 1,742 |
| 2* | 10 | 2,739 | 10 | 2,383 | 10 | 185 |
| 3 | 10 | 4,019 | 10 | 825 | 10 | 1,825 |
β-chemokine production in culture supernatants was assessed on day 5 post-stimulation.
CD4+ and CD8+ cells were obtained by positive selection using immunomagnetic beads rather than by negative selection.
Modulation of HIV Replication in CD4+ and CD8-Depleted PBMC by Endogenous β-Chemokines and Other Proinflammatory Cytokines.
To determine whether endogenous β-chemokines regulate HIV replication in an autocrine/paracrine manner in PBMC of HIV-infected subjects, neutralizing anti-β-chemokine antibodies were added in combination to CD8-depleted PBMC cultured in the absence or the presence of various proportions of CD8+ T cells. Concomitant neutralization of all three β-chemokines failed to rescue HIV replication in CD8-depleted PBMC to which 10% (recall antigen-stimulated cultures; Fig. 2 A and B) or 30% (anti-CD3-stimulated cultures; Fig. 2 C and D) CD8+ T cells had been added. The addition of β-chemokine antibodies to CD8-depleted cultures to which low proportions of CD8+ T cells (<10%) had been added resulted in enhanced HIV replication; however, the degree of enhancement was comparable to that observed in the absence of CD8+ T cells and thus could not be attributed specifically to inhibition of CD8+ T-cell activity (data not shown). Tetanus toxoid is a considerably less potent activator of T cells than is the polyclonal activator anti-CD3, particularly in HIV-infected subjects with reduced capacity to respond to recall antigens. Therefore, the levels of HIV replication and the number of CD8+ cells required to inhibit viral production (Fig. 2 A and B) are lower in tetanus toxoid stimulated conditions as compared with anti-CD3-stimulated cultures (Fig. 2 C and D).
Figure 2.
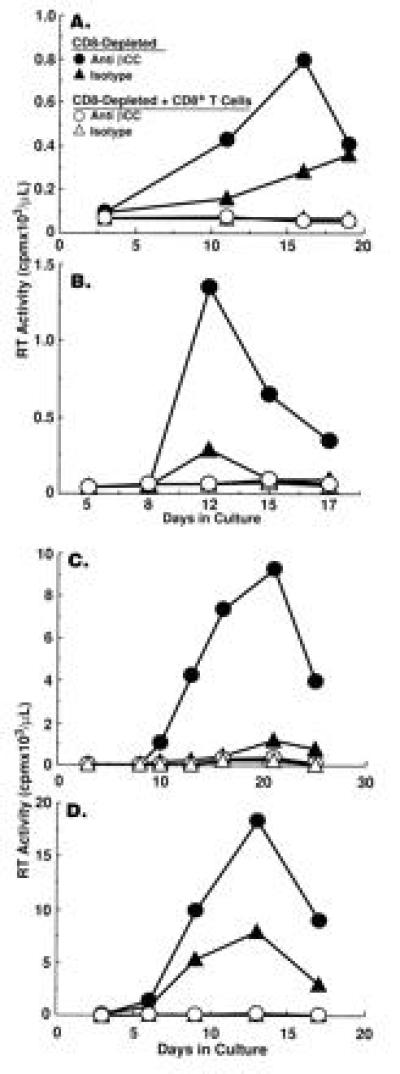
The role of endogenous β-chemokines vs. CD8+ T cells in the regulation of HIV replication in CD8-depleted PBMCs from HIV-infected individuals. CD8-depleted PBMC of four HIV-infected subjects were stimulated with (A and B) tetanus toxoid in the absence or the presence of 10% CD8+ T cells or (C and D) anti-CD3 plus IL-2 in the absence or the presence of 30% CD8+ T cells and treated with either isotype control antibody (110 μg/ml) or a cocktail of anti-β-chemokine antibodies (anti-MIP-1α, 50 μg/ml; anti-MIP-1β, 50 μg/ml; anti-RANTES, 10 μg/ml).
Although neutralization of endogenous β-chemokines failed to rescue HIV replication in the presence of CD8+ T cells, a significant increase in the levels of HIV replication by CD8-depleted PBMC cultured in the absence of CD8+ T cells was observed upon neutralization of endogenous β-chemokines in either recall antigen-stimulated (Fig. 2 A and B) or anti-CD3 plus IL-2-stimulated (Fig. 2 C and D and Fig. 3) conditions in the majority (12, 16) of donors tested. Of interest, the enhancing effect of β-chemokine neutralization on HIV replication was most dramatic in CD8-depleted PBMC cultures from donors in which control levels of in vitro viral production were low as compared with CD8-depleted cultures from donors with high control levels of in vitro HIV replication (Fig. 3); a similar correlation was observed in CD8-depleted PBMC cultures from most of the 16 HIV-infected donors tested. Furthermore, although the numbers of individuals were small (2 of 5 shown in Fig. 3), those with high levels of in vitro viral replication in isotype control cultures had lower CD4+ T-cell counts. The degree of enhancement of HIV replication in CD8-depleted PBMC observed upon neutralization of a particular β-chemokine varied among different donors (Fig. 4) and occasionally enhancement of HIV replication was observed upon neutralization of only one of the β-chemokines (Fig. 4C).
Figure 3.
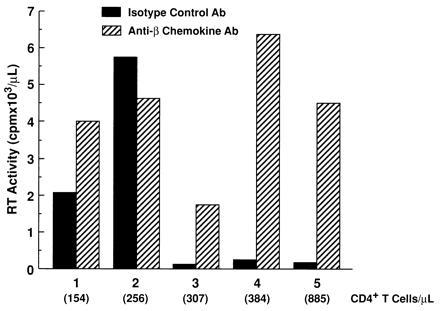
Variable sensitivity of CD8-depleted PBMC of HIV-infected individuals to the enhancing effect on HIV replication by neutralization of β-chemokines. CD8-depleted PBMC from five HIV-infected donors (154–885 CD4+ T cells/μl) were stimulated with anti-CD3 plus IL-2 and cultured in the presence of isotype control antibody or a combination of neutralizing anti-β-chemokine antibodies. Data represent peak HIV replication as measured by reverse transcriptase assay.
Figure 4.
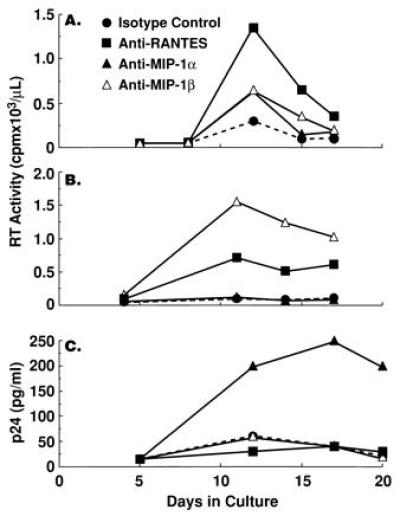
The effect of neutralization of individual endogenous β-chemokines on HIV replication in tetanus toxoid-stimulated CD8-depleted PBMCs from HIV-infected individuals. CD8-depleted PBMCs from three HIV-infected subjects were stimulated with tetanus toxoid in the presence of isotype control antibody (50 μg/ml) or anti-MIP-1α (50 μg/ml), anti-MIP-1β (50 μg/ml), or anti-RANTES (10 μg/ml). Culture supernatants were assayed for the levels of reverse transcriptase activity or p24 antigen at various time points during the culture period.
Several studies have demonstrated that in vitro HIV replication can be inhibited by neutralization of endogenous proinflammatory cytokines, such as TNF-α and IL-1β (7, 16). It was therefore of interest to determine whether the net levels of HIV replication in CD4+ T cells were determined by a balance between the effects of endogenous HIV-inducing proinflammatory cytokines and the HIV-inhibitory β-chemokines. CD4+ PBMC (T cells plus 5% monocytes) from HIV-infected individuals were stimulated with anti-CD3 plus IL-2 in the presence or absence of anti-β-chemokine antibodies, a cocktail of antagonists of proinflammatory cytokines [sTNF receptor (R) plus IL-1 receptor antagonist (ra) plus anti-IL-6] or the two treatments in combination. In CD4+ PBMC of certain HIV-infected donors the levels of HIV replication clearly reflected the balance of positive and negative effects of endogenous proinflammatory cytokines and β-chemokines, respectively (Fig. 5). Cellular proliferation was not significantly altered by any of the treatments despite the dramatic difference in the effects of each treatment on HIV replication (data not shown).
Figure 5.
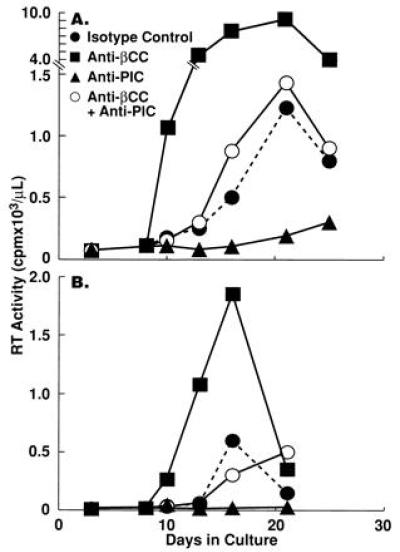
Opposing effects of endogenous β-chemokines and proinflammatory cytokines on HIV replication in CD4+ PBMC of two HIV-infected individuals. CD4+ PBMC (T cells plus 5% monocytes) from HIV-infected subjects were stimulated with anti-CD3 plus IL-2 in the presence of either a cocktail of anti-β-chemokine antibodies (as described in Fig. 3), a cocktail of proinflammatory cytokine antagonists [sTNFR (10 μg/ml), IL-1ra (200 ng/ml), and anti-IL-6 (5 μg/ml)], a combination of both treatments, or isotype control antibodies (100 μg/ml).
DISCUSSION
This study demonstrates that the β-chemokines MIP-1α, MIP-1β, and RANTES inhibit HIV replication in CD8-depleted PBMC of most asymptomatic HIV-infected subjects. It has been well established that β-chemokines are secreted by a variety of cell types including monocytes and a number of lymphocyte subsets (reviewed in ref. 18). We demonstrate that CD4+ T cells from HIV-infected subjects produce β-chemokines at levels comparable to those produced by autologous CD8+ T cells. Of particular interest, neutralization of endogenously produced β-chemokines, individually or in combination, by anti-β-chemokine antibodies, resulted in a significant enhancement of HIV replication in CD8-depleted and CD4+ PBMC from most donors tested; however, these same antibodies failed to eliminate the HIV suppressive effects of CD8+ T cells, when such cells were added in coculture to CD8-depleted PBMCs. Of note is the fact that the level of HIV replication in CD4+ PBMC was found to reflect the net balance of positive and negative regulatory effects of endogenous proinflammatory cytokines (TNF-α, IL-1β, and IL-6) and endogenous β-chemokines, respectively. These data suggest that the β-chemokines, MIP-1α, MIP-1β, and RANTES may play a role in controlling the levels of viral replication in vivo and may counteract or antagonize the effects of HIV-inducing cytokines.
Previous studies from several laboratories have shown that exogenous β-chemokines inhibit the ability of macrophage-tropic HIV strains or cells expressing macrophage-tropic env proteins to acutely infect or fuse with CD4+ T cells and PBMC of normal donors or T-cell lines (15, 16, 17). Our study furthers these observations by demonstrating that the β-chemokines suppress HIV replication in CD8-depleted PBMCs from most asymptomatic HIV-infected subjects stimulated with anti-CD3 plus IL-2 or with recall antigen. Of note is the fact that under the conditions of in vitro antigen stimulation of in vivo infected PBMC, inhibition of HIV replication was often observed at β-chemokine concentrations up to 100-fold less than those previously reported to be required to inhibit HIV replication in acutely infected PHA blasts or T-cell lines (13, 14, 15, 16, 17). It is unclear whether the greater sensitivity to the β-chemokine-mediated HIV inhibition that we observe in our endogenous infection system is due to increased sensitivity of the donor’s HIV quasi-species to inhibition of env binding to CCR-5 by β-chemokines as compared with viruses used in acute in vitro infection systems (15, 16, 17); such strain variability has been noted in previous studies (17). Alternatively, the greater sensitivity could be due to lower levels of CCR-5 on CD4+ cells of the HIV-infected subjects tested or to differences in methodology of this and previous studies (13, 14, 15, 16, 17).
Our observation that simultaneous neutralization of MIP-1α, MIP-1β, and RANTES did not abrogate CD8+ T-cell-mediated HIV suppression is of interest and strongly suggests that the β-chemokines cannot completely explain the HIV suppressor effects of CD8+ T cells, at least in our system of endogenous HIV replication in cells from HIV-infected individuals. However, the assay of suppression of HIV replication conducted in this study on PBMC from HIV-infected subjects used CD8+ T-cell cocultures and not supernatants from CD8+ T cells or human T-lymphocyte virus type I-transformed CD8+ T-cell lines as did the previous study originally describing the HIV-suppressor effects of the β-chemokines (13). Our attempts to conduct these experiments with culture supernatants of primary CD8+ T cells or with transwell coculture systems failed to yield consistent results. In this regard, it is possible that other labile soluble factors or cell contact-mediated factors play a dominant role in suppressing HIV replication in direct coculture systems. However, the observation that negatively selected CD8+ and CD4+ T cells from asymptomatic HIV-infected subjects produce comparable levels of β-chemokines argues against these β-chemokines as the sole mediators of HIV-suppressor activity that is specific for CD8+ T cells. Of particular interest, crosslinking of CD4 molecules by positive selection of CD4+ cells using anti-CD4-coated magnetic beads appeared to reduce the capacity of these cells to produce β-chemokines upon stimulation with PHA (Table 1). This observation suggests that antibody crosslinking of CD4 on the surface of T cells, as seen here, or by glycoprotein 120/160 in vivo, may deliver a negative signal with regard to the production of β-chemokines.
The role of endogenous β-chemokines produced by PBMC subsets other than CD8+ T cells in the regulation of HIV replication in CD4+ T cells from HIV-infected subjects has not been previously demonstrated. Previous studies (14) suggested that elevated β-chemokine production by CD4+ cells from exposed uninfected individuals may be responsible for the lack of susceptibility to acute infection with a primary isolate of HIV, but not a variant of this strain modified to express a T-cell tropic-like env gene product; this observation was later found to be due to the lack of CCR-5 expression in these exposed uninfected individuals (20). Data from our β-chemokine neutralization studies suggest that high levels of the natural ligands of CCR-5, MIP-1β, MIP-1α, and RANTES, in HIV-infected subjects, can play a significant role in limiting the spread of HIV infection in most asymptomatic HIV-infected individuals. We have presumed that the individuals in this study were harboring predominantly macrophage-tropic strains because, for the most part, they were in the early stages of HIV disease (21). The variability in the capacity of anti-β-chemokine antibodies to enhance in vitro HIV replication in CD8-depleted or CD4+ PBMC from HIV-infected individuals may be a reflection of the relative representation of T-cell-tropic vs. macrophage-tropic viruses in the PBMCs of the subjects under study. In this regard, neutralization of endogenous β-chemokines appeared to have a more consistent and dramatic enhancing effect on viral replication in those CD8-depleted PBMCs from donors with higher numbers of CD4+ T cells/μl and in whose cultures the control levels of in vitro HIV production were moderate-to-low (Fig. 3). Although the reasons for these observations are unclear at present they are consistent with the hypothesis that the predominant virus(es) in individuals with early-to-intermediate stage disease are slow/low, macrophage tropic strains (21). Correlations of the type of virus obtained from PBMC cultures with the suppressive capabilities of the endogenous β-chemokines in these cultures are currently under investigation in our laboratory. Alternatively, the inability of anti-β-chemokine antibodies to enhance HIV replication in CD8-depleted PBMC cultures from certain individuals could be due to lack of or a great reduction of β-chemokine production; however, based on our measurements of β-chemokine levels, this does not appear to be the case.
The fact that endogenous proinflammatory cytokines are important modulators of HIV replication in primary PBMCs has been previously demonstrated (reviewed in refs. 1, 3, 7, and 22). The enhancing effect of proinflammatory cytokines has been demonstrated in culture systems employing both T-cell-tropic (7, 16) and macrophage-tropic (3, 7, 16) strains of HIV. It is interesting to consider the selective pressures exerted by proinflammatory cytokines and β-chemokines on the emergence of predominantly T-cell-tropic, rapid/high virus strains in HIV-infected individuals during disease progression. In this regard, both the β-chemokines and the HIV-inducing cytokines TNF-α, IL-1β, and IL-6 are involved in primary proinflammatory immune responses (23) and have been reported to be produced at elevated levels in HIV-infected individuals, as determined either by in situ tissue analysis or plasma levels (24, 25, 26, 27, 28, 29). Taken together, these data strongly suggest that the steady state of virus replication in HIV-infected individuals reflects, at least in part, a delicate balance between cytokines that upregulate and down-regulate HIV replication. Although several of these cytokines have already been identified, it is highly likely that more will be discovered.
Acknowledgments
We would like to thank Patricia Walsh for her excellent editorial assistance and Joe Adelsberger for performing flow cytometry analyses. A.L.K. performed this project in partial fulfillment of the requirements of the Ph.D. program of the Department of Microbiology and Immunology at George Washington University (Washington, DC). A.O. is the recipient of a fellowship for AIDS research from the Instituto Superiore di Sanita (Rome).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: CCR-5, chemokine receptor 5; PBMC, peripheral blood mononuclear cells; MIP-1α, MIP-1β, macrophage inflammatory proteins 1α and 1β; RANTES, regulated on activation normally T-cell expressed and secreted; IL, interleukin; TNF, tumor necrosis factor; rh, recombinant human; TT, tetanus toxoid; PHA, phytohemagglutinin.
References
- 1.Poli G, Fauci A S. In: Human Cytokines: Their Role in Disease and Therapy. Aggarwal B B, Puri R K, editors. Cambridge, MA: Blackwell Scientific; 1995. pp. 421–449. [Google Scholar]
- 2.Ho D D, Hartshorn K L, Rota T R, Andrews C A, Kaplan J C, Schooley R T, Hirsch M S. Lancet. 1985;i:602–604. doi: 10.1016/s0140-6736(85)92144-0. [DOI] [PubMed] [Google Scholar]
- 3.Weissman D, Poli G, Fauci A S. AIDS Res Hum Retroviruses. 1994;10:1199–1206. doi: 10.1089/aid.1994.10.1199. [DOI] [PubMed] [Google Scholar]
- 4.Akridge R E, Oyafuso L K, Reed S G. J Immunol. 1994;153:5782–5789. [PubMed] [Google Scholar]
- 5.Osborn L, Kunkel S, Nabel G J. Proc Natl Acad Sci USA. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poli G, Kinter A L, Justement J S, Kehrl J H, Bressler P, Stanley S, Fauci A S. Proc Natl Acad Sci USA. 1990;87:782–785. doi: 10.1073/pnas.87.2.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kinter A L, Poli G, Fox L, Hardy E, Fauci A S. J Immunol. 1995;154:2448–2459. [PubMed] [Google Scholar]
- 8.Folks T M, Justement J S, Kinter A L, Dinarello C A, Fauci A S. Science. 1987;238:800–802. doi: 10.1126/science.3313729. [DOI] [PubMed] [Google Scholar]
- 9.Poli G, Kinter A L, Fauci A S. Proc Natl Acad Sci USA. 1994;91:108–112. doi: 10.1073/pnas.91.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuitemaker H, Kootstra N A, Koppelman M H, Bruisten S M, Huisman H G, Tersmette M, Miedema F. J Clin Invest. 1992;89:1154–1160. doi: 10.1172/JCI115697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poli G, Bressler P, Kinter A L, Duh E, Timmer W C, Rabson A, Justement J S, Stanley S, Fauci A S. J Exp Med. 1990;172:151–158. doi: 10.1084/jem.172.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poli G, Kinter A L, Justement J S, Bressler P, Kehrl J H, Fauci A S. J Exp Med. 1991;173:589–597. doi: 10.1084/jem.173.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cocchi F, DeVico A L, Garzino-Demao A, Arya S K, Gallo R C, Lusso P. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 14.Paxton W A, Martin S R, Tse D, O’Brien T R, Skurnick J, VanDevanter N L, Padian N, Braun J F, Kotler D P, Wolinsky S M, Koup R. Nat Med. 1996;2:412–417. doi: 10.1038/nm0496-412. [DOI] [PubMed] [Google Scholar]
- 15.Alkhatib G, Combadiere C, Broder C C, Feng Y, Kennedy P E, Murphy P M, Berger E A. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 16.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton R E, Hill C M, Davis C B, Peiper S. Nature (London) 1996;381:661–667. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 17.Dragic T, Litwin V, Allaway G P, Martin S R, Huang Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. Nature (London) 1996;361:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 18.Taub D D, Oppenheim J J. Ther Immunol. 1994;1:229–246. [PubMed] [Google Scholar]
- 19.Willey R L, Smith D H, Lasky L A, Theodore T S, Earl P L, Moss B, Capon D J, Martin M A. J Virol. 1988;62:139–147. doi: 10.1128/jvi.62.1.139-147.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu R, Paxton W A, Choe S, Ceradini D, Martin S R, Horuk R, MacDonald M E, Stuhlmann H, Koup R A, Landau N R. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 21.Schuitemaker H, Koot M, Kootstra N A, Dercksen M W, de Goede R E, van Steenwijk R P, Lange J M, Schattenkerk J K, Miedema F, Tersmette M. J Virol. 1992;66:1354–1360. doi: 10.1128/jvi.66.3.1354-1360.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vyakarnam A, McKeating J, Meager A, Beverley P C. AIDS. 1990;4:21–27. doi: 10.1097/00002030-199001000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Ben-Baruch A, Michiel D F, Oppenheim J J. J Biol Chem. 1995;270:11703–11706. doi: 10.1074/jbc.270.20.11703. [DOI] [PubMed] [Google Scholar]
- 24.Denis M, Ghadirian E. Clin Exp Immunol. 1994;96:187–192. doi: 10.1111/j.1365-2249.1994.tb06540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidtmayerova H, Nottet H S, Nuovo G, Raabe T, Flanagan C R, Dubrovsky L, Gendelman H E, Cerami A, Bukrinsky M, Sherry B. Proc Natl Acad Sci USA. 1996;93:700–704. doi: 10.1073/pnas.93.2.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tedla N, Palladinetti P, Kelly M, Kumar R K, DiGirolamo N, Chattophadhay U, Cooke B, Truskett P, Dwyer J, Wakefield D, Lloyd A. Am J Pathol. 1996;148:1367–1373. [PMC free article] [PubMed] [Google Scholar]
- 27.Lahdevirta J, Maury C P, Teppo A M, Repo H. Am J Med. 1988;85:289–291. doi: 10.1016/0002-9343(88)90576-1. [DOI] [PubMed] [Google Scholar]
- 28.Emilie D, Peuchmaur M, Maillot M C, Crevon M C, Brousse N, Delfraissy J F, Dormont J, Galanaud P. J Clin Invest. 1990;86:148–159. doi: 10.1172/JCI114678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott-Algara D, Vuillier F, Marasescu M, Saint Martin D E, Dighiero G. AIDS Res Hum Retroviruses. 1991;7:381–386. doi: 10.1089/aid.1991.7.381. [DOI] [PubMed] [Google Scholar]