

Abstract
Previous work has suggested that arsenic exposure contributes to skin carcinogenesis by preserving the proliferative potential of human epidermal keratinocytes, thereby slowing the exit of putative target stem cells into the differentiation pathway. To find a molecular basis for this action, present work has explored the influence of arsenite on keratinocyte responses to epidermal growth factor (EGF). The ability of cultured keratinocytes to found colonies upon passaging several days after confluence was preserved by arsenite and EGF in an additive fashion, but neither was effective when the receptor tyrosine kinase activity was inhibited. Arsenite prevented the loss of EGF receptor protein and phosphorylation of tyrosine 1173, preserving its capability to signal. The level of nuclear β-catenin was higher in cells treated with arsenite and EGF in parallel to elevated colony forming ability, and expression of a dominant negative β-catenin suppressed the increase in both colony forming ability and yield of putative stem cells induced by arsenite and EGF. As judged by expression of three genes regulated by β-catenin, this transcription factor had substantially higher activity in the arsenite/EGF-treated cells. Trivalent antimony exhibited the same effects as arsenite. A novel finding is that insulin in the medium induced the loss of EGF receptor protein, which was largely prevented by arsenite exposure.
Keywords: Antimony, Arsenite, β-Catenin, Epidermal growth factor, Insulin, Keratinocyte
Introduction
Human populations chronically exposed to high levels of arsenic in drinking water suffer a variety of health problems. Among the observed afflictions, skin lesions are common, with hyperkeratosis developing after a few years of exposure (Verret et al, 2005) followed by development of squamous and basal cell carcinomas (Guo et al, 2001 ). Although arsenic is an established human carcinogen based on epidemiological data, administration of arsenic alone has not been found to cause cancer in animal models except for mice treated in utero (Waalkes et al, 2004). However, arsenic is a tumor co-promoter in mouse epidermis expressing the v-ras oncogene (Germolec et al, 1998) and a skin co-carcinogen with UV light (Burns et al, 2004). Careful examination of epidemiological data in Bangladesh indicates sun exposure is a risk factor for arsenical keratoses, precursors of skin cancer (Chen et al, 2006). Among the many known cellular effects of arsenic that may contribute to its action as a co-carcinogen or co-promoter (Kitchin, 2001), including DNA damage (Hei and Filipic, 2004), one explored in present work is its preservation of keratinocyte proliferative potential, a feature demonstrable in culture (Patterson et al, 2005).
Human keratinocytes cultured with 3T3 feeder layer support provide a useful model for studying effects of toxic agents and carcinogens. At confluence, the cells form a stratified epithelium with many of the features exhibited by natural epidermis (Green, 1979). Basal cells leave the germinative pool to undergo the process of terminal differentiation, while superficial cells express differentiation markers, form cornified envelopes and lose the ability to generate colonies when replated. Potential stem cell markers, enriched in the pool of proliferative keratinocytes, include β1-integrin and β-catenin (Zhu and Watt, 1999). Subpopulations of epidermal keratinocytes with differing proliferative capacities (Barrandon and Green, 1987) are enriched in β1-integrin and are separable by their rates of adhesion to the extracellular matrix components collagen IV and fibronectin (Jones and Watt, 1993). Arsenite has been found to prevent the decline of β1-integrin and transcriptionally active β-catenin in cultured normal and spontaneously immortalized human epidermal keratinocytes and thus to preserve the fraction of rapidly adhering cells with high proliferative potential (Patterson et al, 2005).
Finding the mechanism by which arsenite maintains keratinocyte proliferative potential in culture, resulting in enrichment of putative stem cells, likely will help elucidate its carcinogenic action. To this end, examining its influence on signaling by growth factors such as EGF and insulin promises to be helpful. EGF has long been known to increase clonal expansion, culture lifespan and growth potential of human epidermal keratinocytes (Barrandon and Green, 1987; Rheinwald and Green, 1977), and excessive expression of the EGFR ligand transforming growth factor-α sensitizes mouse skin to the tumorigenic action of polycyclic aromatic hydrocarbons and activated oncogenes (Shibata et al, 1997; Wang et al, 1994). The EGFR and other ErbB family members are overexpressed in various tumor types, contributing to development of neoplasia. This phenomenon has stimulated much effort to elucidate normal and aberrant EGFR regulatory processes and possible pharmaceutical intervention (Sweeney and Carraway, 2004). The finding that hydrogen peroxide activates the EGFR while preventing its c-Cbl mediated down-regulation (Ravid et al, 2002) has raised the possibility that other exogenous agents producing oxidative stress, such as arsenite, may have similar effects.
Insulin, present at high levels in the culture medium, signals through the insulin and IGF-1 receptors. These receptors share elements of their signaling pathways but display differences in downstream effects (Shen et al, 2001). As shown by gene ablation studies in mice, signaling through the insulin receptor and, more profoundly, IGF-1R are important for the proper development and function of epidermis. The epidermis in mice lacking IGF-1R function is thinner than normal (Liu et al, 1993), and the keratinocytes in culture display decreased proliferation and accelerated differentiation (Sadagurski et al, 2006). Consistent with observations that IGF-1 or IGF-1R are overexpressed in many human tumors, overexpression of IGF-1 in mouse epidermal basal cells acts as a tumor promoting stimulus (DiGiovanni et al, 2000). Moreover, cross-talk between the EGFR and IGF-1R has been observed to have a significant effect on signaling (Adams et al, 2004), where IGF-1R ligands can activate the EGFR (Knowlden et al, 2005). By contrast, the skin of mice lacking the insulin receptor is grossly normal, although an obvious histological abnormality is the expression of keratin 6 throughout the spinous layer (Wertheimer et al, 2001).
The current study addresses the roles of EGFR and IGF-1R/IR in regulation of keratinocyte proliferative potential and their modulation by arsenite. Transcriptionally active, nuclear β-catenin is demonstrated to be required for the increased proliferative potential elicited by arsenite in an EGFR-dependent manner. Contrary to expectation, insulin is shown to decrease levels of EGFR and active β-catenin. We propose that arsenite treatment counteracts the effects of insulin by preventing degradation of the EGFR, resulting in prolonged signaling and, ultimately, stabilization of β-catenin and preservation of the putative stem cell population. Beyond its relevance for carcinogenesis, arsenic antagonism of insulin action, as previously reported in adipocyte glucose uptake, is of particular interest in view of its possible contribution to understanding the mechanism of arsenic-induced diabetes (Walton et al, 2004).
Materials and methods
Cell culture
Spontaneously immortalized (SIK) (Rice et al, 1993) and normal human epidermal cells (hEp) derived from foreskin were propagated in DMEM/F12 (2:1) medium supplemented with fetal bovine serum (5%), hydrocortisone (0.4 μg/ml), EGF (10 ng/ml), adenine (0.18 mM), insulin (5 μg/ml except where noted), transferrin (5 μg/ml) and triiodothyronine (20 pM), using a feeder layer of lethally irradiated 3T3 cells (Allen-Hoffmann and Rheinwald, 1984). Medium was further supplemented with cholera toxin (10 ng/ml) at inoculation, replaced with EGF (10 ng/ml) starting at the first medium change. Cells were grown until just before confluence with medium changes at 3 to 4 day intervals, at which time they were treated with 2 μM sodium arsenite, 5 μM antimony tartrate, 10 ng/ml EGF, or switched to medium without insulin. Although the cells grew less vigorously in the absence of insulin, its removal at confluence largely prevented the marked decline in colony forming ability noted previously (Patterson et al, 2005). In certain instances, cells were first pre-treated for 1 h with 1 μM AG1478 (LC Laboratories, Woburn, MA), 1 μM PD 158780 (Calbiochem, San Diego, CA), 2 μg/ml mouse monoclonal EGFR antibody (clone 225, Calbiochem), or 30 μg/ml cycloheximide before the indicated treatment.
Retroviral infection of SIK
The cDNA of the β-catenin T3 dominant negative deletion mutant (Funayama et al, 1995) was subcloned into the retroviral vector pBabe-puro. A pBabe-puro vector only, used as control, or the T3 construct were transfected into the retroviral packaging cell line Phoenix Ampho. The retrovirus collected from the medium of Phoenix Ampho transfected cultures was then transferred to SIK cultures in the presence of 4 μg/ml polybrene. Infected cells were selected using 1 μg/ml puromycin. To confirm expression of the T3 in infected cells, protein extracts were probed with a hemagglutin antibody to detect HA-tagged β-catenin, yielding a band of 50 kDa, which was absent in SIK cultures infected with vector alone. Expression of the HA-tag was observed immunocytochemically in 80–90% of the cells. SIK infected with the T3 construct had slower growth rates and reduction of CFE to 30–40% of cells infected with vector alone (data not shown).
Colony forming efficiency (CFE), rapidly adhering (RAC), and slowly adhering (SAC) cell assays
CFE, RAC and SAC assays were performed as previously described (Jones and Watt, 1993; Patterson et al, 2005). Briefly, CFE was determined by treating cultures for indicated times, trypsinizing them, and inoculating 103 or 104 cells in 6 cm dishes. Grown with feeder layer support until the majority were 2–5 mm in diameter, the colonies were fixed and visualized by staining with Rhodanile blue (Rheinwald and Green, 1975). For RAC and SAC assays, cells were fractionated by their rates of adherence to dishes coated with collagen IV. Dishes (6 cm) were coated with 1.2 ml of 50 μg/ml collagen type IV, incubated overnight at 4°C and then blocked with 0.5 mg/ml heat denatured BSA for 1 hr. Cells were placed on coated dishes for 15 min (RACs). The unattached cells were removed and allowed to attach to a second set of coated dishes for 45 min (SACs). After the attachment period, cultures were rinsed with serum free medium, 3T3 feeder cells were added and the keratinocytes were allowed to grow until the majority of the colonies were at least 2 mm in diameter, typically one to two weeks.
Immunnoblot analysis
For most experiments, cells were scraped directly into buffer containing 2% SDS, 62.5 mM Tris (pH 6.8) and 10% glycerol. Protein was measured with bicinchoninic acid (Pierce) before addition of DTT to 50 mM. In a given experiment, equal amounts (20 μg) of protein were submitted to SDS polyacrylamide gel electrophoresis, transferred to PVDF membranes, blocked with 5% dry milk in TBST, incubated with the indicated antibodies and detected using ECL Plus chemiluminescence detection reagent (GE Healthcare/Amersham, Piscataway, NJ). Antibodies were obtained from the following sources: β-catenin (rabbit polyclonal) and EGFR (rabbit polyclonal) from Cell Signaling Technology (Danvers, MA); hemagglutin (clone HA.11) from Covance (Berkeley, CA); actin (clone AC-74) from Sigma (St. Louis, MO); ABC β-catenin (clone 8E7), which measures β-catenin dephosphorylated on serine 37 and threonine 41 residues, from Upstate/Millipore (Temecula, CA); phosphorylated tyrosine 1173 EGFR (rabbit polyclonal) and E-cadherin (clone G-10) from Santa Cruz Biotechnology (Santa Cruz, CA).
Ras activation
Measurement of active Ras was performed using a Ras Activation Kit (Upstate/Millipore, Temecula, CA). Briefly, cultures were harvested in buffer supplemented with 1 mM β-glycerophosphate, 0.2 mM sodium vandate and EDTA-free protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Homogenates were clarified by microfuge centrifugation, incubated with Raf-1 RBD agarose beads, electrophoresed in acrylamide gels and blotted with monoclonal antibody RAS10.
Preparation of nuclear extracts for β-catenin immunoblotting
Cells were scraped from 10 cm dishes into ice-cold PBS, recovered by low speed centrifugation and resuspended in buffer containing 0.1 M HEPES (pH 7.4), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 10 mM β-glycerophosphate, 10 μM sodium vanadate, 1 mM PMSF and protease inhibitor cocktail (Roche, Nutley, NJ). The cells were disrupted by Dounce homogenization and crude nuclei were prepared by centrifugation at 3300xg for 15 min. Nonhistone nuclear proteins were extracted in 0.43 M NaCl – 1 mM EDTA –20 mM Hepes buffer (pH 7.4) for 30 min at 4°C and recovered by centrifugation at 100,000xg for one h at 4°C. Supernatants were confirmed to be free of E-cadherin by immunoblotting.
Real-time reverse transcription PCR
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA). To avoid amplification of genomic DNA, RNA was pretreated with DNase (DNA-free kit from Ambion/Applied Biosystems, Foster City, CA). c-DNA synthesis was performed using High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). The cDNA served as a template in quantitative real-time PCR utilizing TaqMan Fast Universal PCR Master Mix and TaqMan Gene Expression assay probes for the EGFR, MMP-7, PPARδ, BMP-4 or18s RNA (Applied Biosystems), and an ABI 7500 Fast Sequence Detection System. mRNA expression, normalized to endogenous 18S RNA, is presented relative to untreated control cultures in the presence of insulin (set to 1.0).
Results
Preservation of proliferative potential by treatment with arsenite or EGF or by insulin removal
The proliferative potential of epidermal keratinocytes, measured by CFE, decreased substantially when the cultures were held at confluence under standard culture conditions (Patterson et al, 2005). In the present work, the CFE of SIK cultures decreased 80% by three days after confluence and 90% by 9 days as the cells stratified, exited the proliferative compartment and differentiated. This loss was largely prevented if the cultures were treated with arsenite and EGF or if the insulin in the medium was removed. As seen in Fig. 1A, cells cultured for three days after confluence in the presence of 2 μM arsenite or 10 ng/ml EGF exhibited approximately double the CFE of untreated cultures, while those treated with both agents had four fold the CFE. Antimonite (SbIII) was as effective as arsenite, and co-treatment with EGF was again also additive. Removing insulin from the medium at confluence also increased CFE. By contrast, addition of EGF (Fig. 1A) or arsenite (not shown) did not further increase CFE of cultures grown after confluence in the absence of insulin. Similar results were obtained 9 days after confluence with one clear difference. At this later time point, EGF alone had little effect, but it still augmented the action of arsenite to yield the same CFE as with cultures incubated in the absence of insulin. At the 3 day time point, results with hEp were comparable to those with SIK cultures (Fig. 1B). However, addition of arsenite and EGF together was more powerful than simply omitting insulin. Moreover, in the absence of insulin, EGF substantially augmented CFE (Fig 1C), while arsenite was ineffective (data not shown).
Fig. 1.
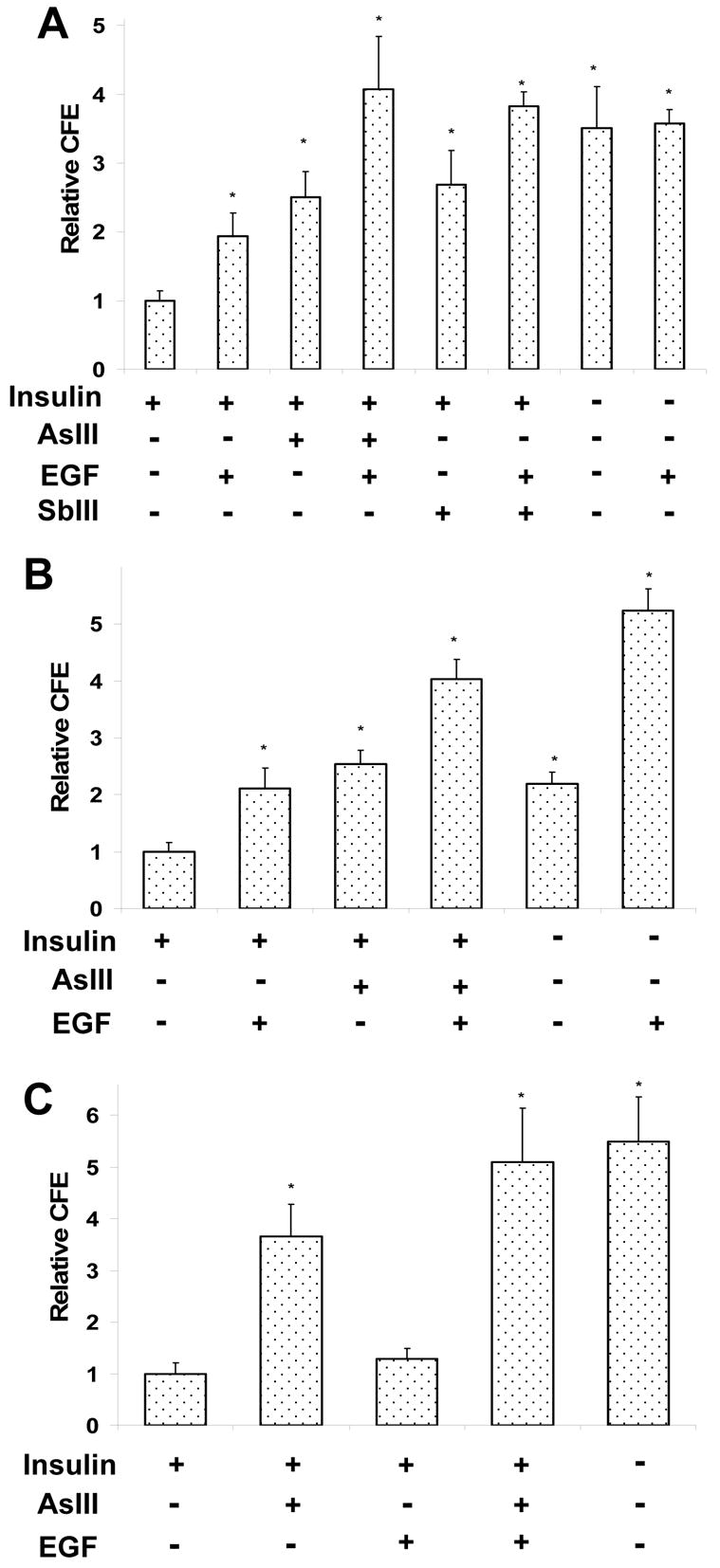
Preservation of proliferative potential by arsenic, antiomony and EGF. Starting at 90% confluence, SIK (A,C) or hEp (B) cultures were treated with 2 μM arsenite, insulin-free medium or 5 μM antimonite with and without 10 ng/ml EGF for 3 days (A,B) or 9 days (C) before measuring colony forming efficiencies (CFE). Values are relative to CFE of untreated cells in medium containing insulin, which is set as 1. Student’s t-test showing values significantly different from untreated cultures (*) with Bonferroni correction gave p<0.005 from averages of 4 (A), 2 (B) or 3 (C) independent experiments performed in duplicate.
Arsenite, insulin, and antimonite effects mediated through the EGFR
In the presence of the EGFR inhibitor AG1478, neither arsenite nor antimonite increased CFE in either SIK (Fig. 2A) or hEp cultures (not shown) regardless of the addition of EGF. As seen, AG1478 did not affect CFE in the presence of insulin, but largely prevented the increase in CFE resulting from insulin removal. The structurally unrelated EGFR inhibitor, PD 158780 at 1 μM gave similar results. Arsenite treatment increased active Ras, a downstream effector of the EGFR, whereas co-treatment with AG1478 prevented Ras activation by arsenite (Fig. 2B).
Fig. 2.
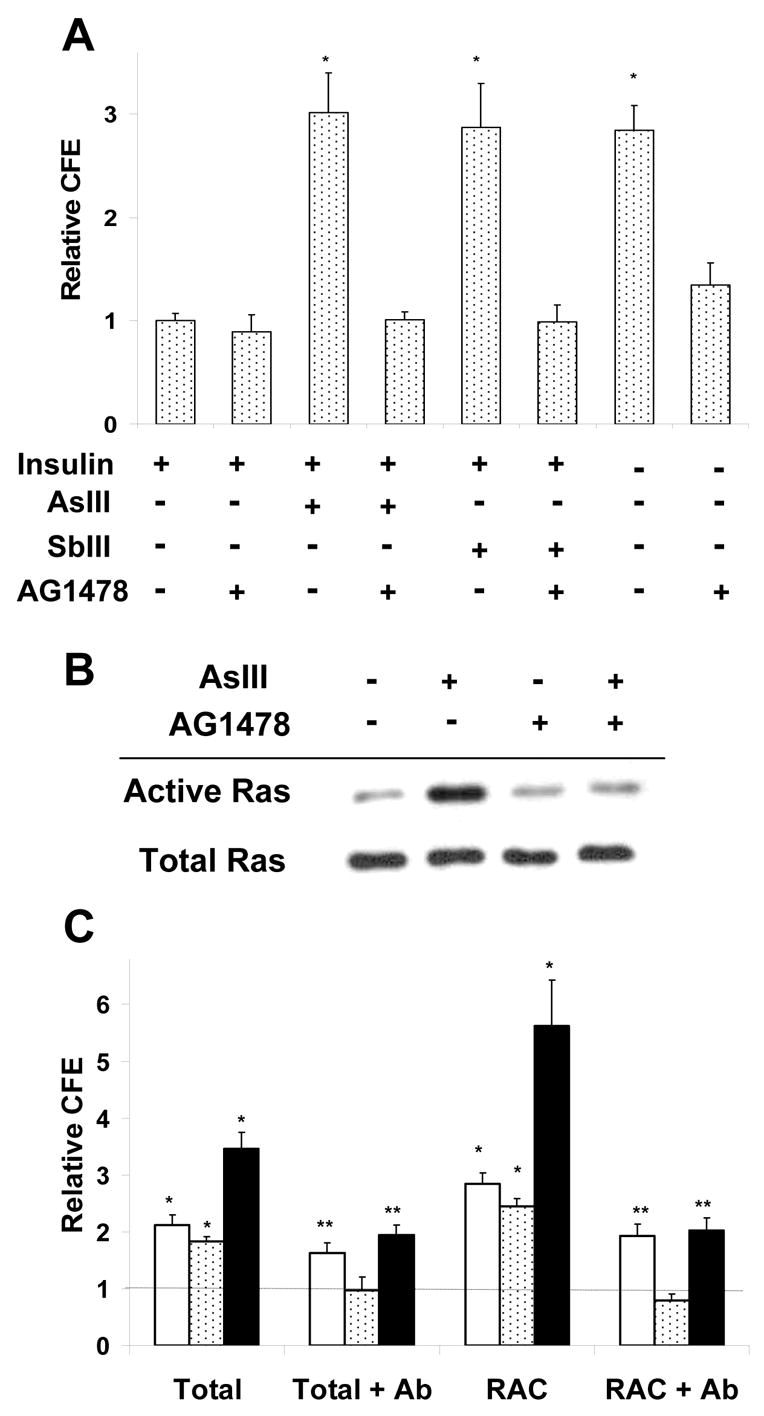
Proliferative potential of SIK cultures treated for 3 days starting at 90% confluence with arsenite, antimonite, AG1478 or without insulin. (A) Treatments were as indicated in medium containing EGF. CFE values are relative to those of cultures in the presence of insulin, set at 1; Student’s t-test with Bonferroni correction (*, p<0.006) calculated from 5 (arsenite) or three (antimonite and no insulin) experiments performed in duplicate. (B) After 3 days treatment as indicated in the presence of insulin, active Ras was detected by pull down assay. Levels of Ras in total cell lysates, probed in parallel, were unchanged. Blots are representative of three independent experiments. (C) Cells were pretreated for one hr with 8 μg of C-225 EGFR antibody before addition of 2 μM arsenite (clear bars), 10 ng/ml EGF (stippled bars) or arsenite and EGF (solid bars). After 3 days, CFE of unfractionated and rapidly adhering cells (RAC) were measured. Illustrated are CFEs relative to untreated controls, indicated by the horizontal line at 1. Values significantly different from the control (*) were judged by Student’s t-test with Bonferroni correction (*, p<0.004; **, P<0.01).
Preventing EGF binding to the EGFR by addition of an EGFR neutralizing antibody to the medium greatly attenuated the increase in CFE resulting from EGF (but not arsenite) treatment (Fig. 2C) or insulin removal (not shown). It also reversed the additive effect of co-treating with both arsenite and EGF, resulting in a CFE similar to that with arsenite treatment alone (Fig. 2C). Since arsenite has its greatest effect on the proliferative capacity of RACs (Patterson et al, 2005), this population was isolated for analysis. As in the unfractionated population, the EGFR neutralizing antibody prevented increased CFE due to addition of EGF, but not of arsenite (Fig. 2C).
EGFR stability and signaling after treatment with arsenite, EGF or insulin
In untreated cultures EGFR protein levels decreased after confluence as cells exited the proliferative pool and differentiated. Arsenite or antimonite treatment prevented this decrease, preserving total EGFR levels starting as early as one day after treatment (Fig. 3A). EGFR phosphorylation on tyrosine 1173, an indicator of activity, was considerably higher in cultures treated with arsenite (Fig. 3B). However, unlike EGF, which increased tyrosine phosphorylation quickly but transiently, arsenite treatment resulted in a higher relative level of phosphorylation that persisted for the duration of treatment and did not depend on the presence of EGF. Insulin suppressed EGFR levels in a dose-dependent fashion, with dramatically higher levels evident with no added insulin (Fig. 3C). Further, insulin suppressed relative EGFR tyrosine 1173 phosphorylation, indicative of reduced activity.
Fig. 3.
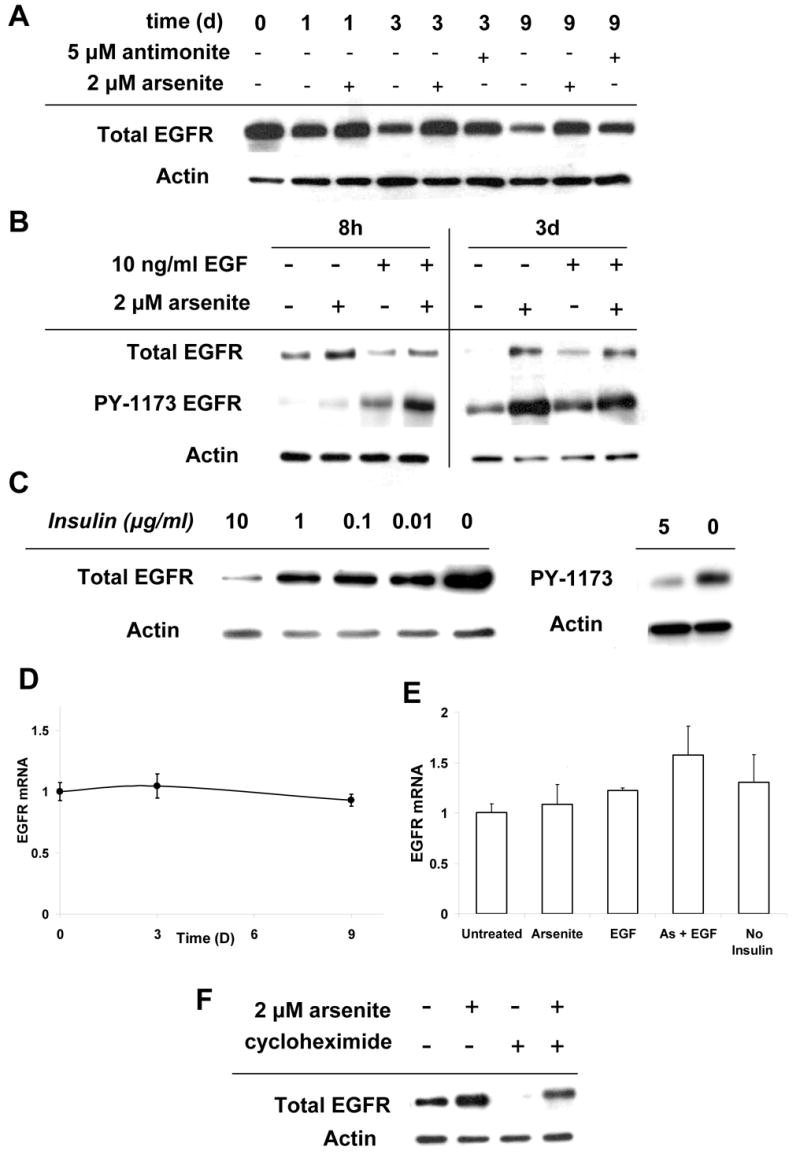
EGFR levels and activity in response to arsenite or insulin. (A) SIK cultures were treated as indicated starting at 90% confluence (day 0) and processed for immunoblot analysis on the indicated day. (B) Cells were treated as indicated for 8 hr (left) or 3 days (right) and probed with antibodies to total EGFR or EGFR phosphorylated at tyrosine 1173 (PY-1173). (C) Immunoblot of total EGFR protein (left) and PY-1173 (right) after treatment with indicated concentrations of insulin for 6 days (left) or 3 days (right). (D,E) EGFR mRNA expression was measured using real time PCR in SIK cultures treated as indicated (3 days in E). (F) SIK cultures untreated or pretreated for one hr with 30 μM cycloheximide were exposed to 2 μM arsenite for 24 hr and then harvested for immunoblot analysis. β-actin was used as loading control.
To determine whether differences in EGFR levels were due to altered gene expression, EGFR mRNA levels were quantitated by real-time PCR in samples harvested after three days of treatment. Little change in gene expression was observed among untreated cultures from days 0 to 9 (Fig. 3D) or those treated with arsenite in the presence or absence of EGF or incubated after confluence in the absence of insulin (Fig. 3E). To determine whether arsenite prevented degradation of the EGFR, protein synthesis was arrested with cycloheximide. During the course of a day, the EGFR was completely depleted in cells treated with EGF, whereas arsenite treatment prevented most of the degradation (Fig. 3F).
Arsenite or antimonite addition or insulin removal stabilize nuclear β-catenin
As previously demonstrated, cultures treated with arsenite exhibited elevated levels of nuclear (transcriptionally active) β-catenin compared to untreated ones (Patterson et al, 2005). In present work, β-catenin dephosphorylated at serine 37 and threonine 41 (ABC β-catenin, transcriptionally active and resistant to degradation by the APC destruction complex) was elevated after arsenite treatment. Like EGFR, active β-catenin decreased after confluence, a phenomenon prevented with arsenite or antimonite treatment (Fig. 4A). As seen in Fig. 4B, arsenite or EGF elevated active β-catenin, while co-treatment resulted in an approximately additive increase. As shown, increased active β-catenin was reflected in an increased nuclear level. Removal of insulin from the medium also led to higher levels of active β-catenin (Fig. 4D).
Fig. 4.
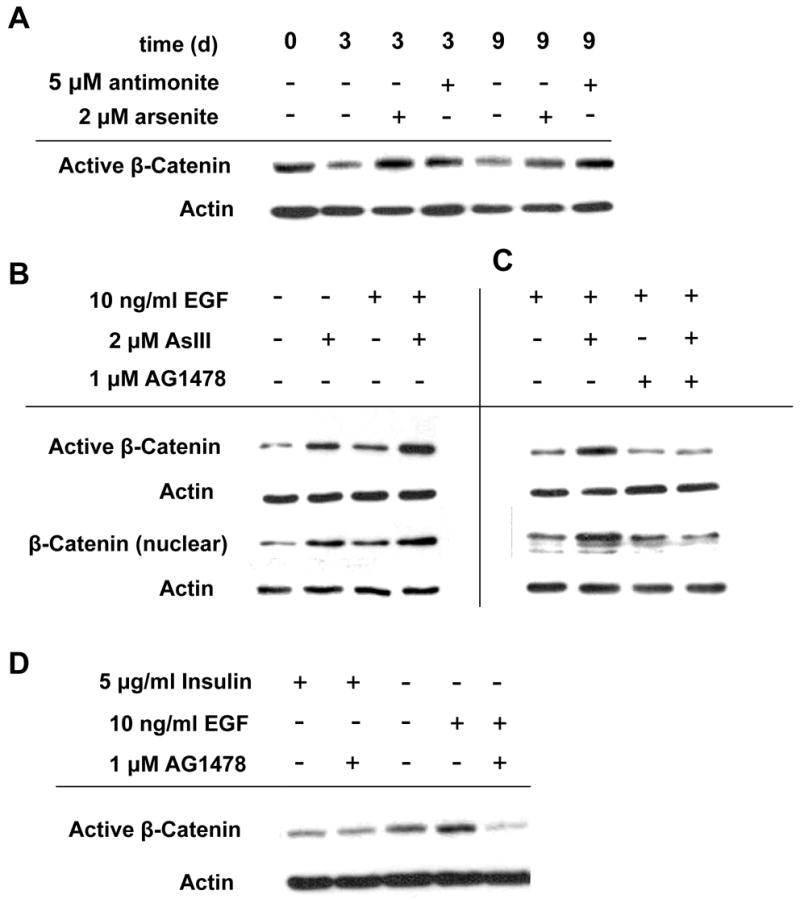
Stabilization of β-catenin protein levels by treatment with arsenite, antimonite or in the absence of insulin depends upon EGFR activation. (A) SIK cultures were treated as indicated starting at 90% confluence (day 0) followed by immunoblot analysis using ABC β-catenin, an antibody that detects the active, non-phosphorylated form. (B,C) SIK cultures were treated for 3 dyas with the indicated treatment followed by immunoblot analysis of total cell lysates with ABC β-catenin antibody or nuclear extracts with total β-catenin antibody. (D) Active β-catenin levels in hEp after 3 days of indicated treatment. β-Actin was used as a loading control.
Since both EGFR and β-catenin were required for arsenite preservation of CFE, we asked whether inhibition of EGFR prevented stabilization of active β-catenin. In the presence of the EGFR inhibitor AG1478 (Fig. 4C) or PD158780 (not shown), arsenite increased neither nuclear nor active β-catenin. Pre-treatment with AG1478 also prevented the persistence of active β-catenin in the absence of insulin (Fig. 4D).
To test directly the requirement for active β-catenin by agents that increase CFE, SIK cultures were retrovirally infected with a dominant negative T3 β-catenin construct (Funayama et al, 1995) or, as a control, with the pBabe-puro vector without the T3 insert. In cells infected with vector only, treatment with arsenite (or antimonite) for 3 days increased CFE in the unfractionated population (Fig. 5A) as well as in populations of RACs and SACs (Fig. 5B,C). Cells expressing the dominant negative β-catenin were not responsive to arsenite- or antimonite-induced increases in CFE of unfractionated and RAC populations (Fig. 5A,B) and were only partially responsive to EGF and insulin removal (Fig. 5A). T3 expression reduced, but did not completely eliminate, increased CFE due to co-treatment with arsenite and EGF, resulting in a CFE similar to that obtained with EGF alone (Fig. 5A,B). The CFE of the SAC population was much less affected by T3 expression (Fig. 5C).
Fig. 5.
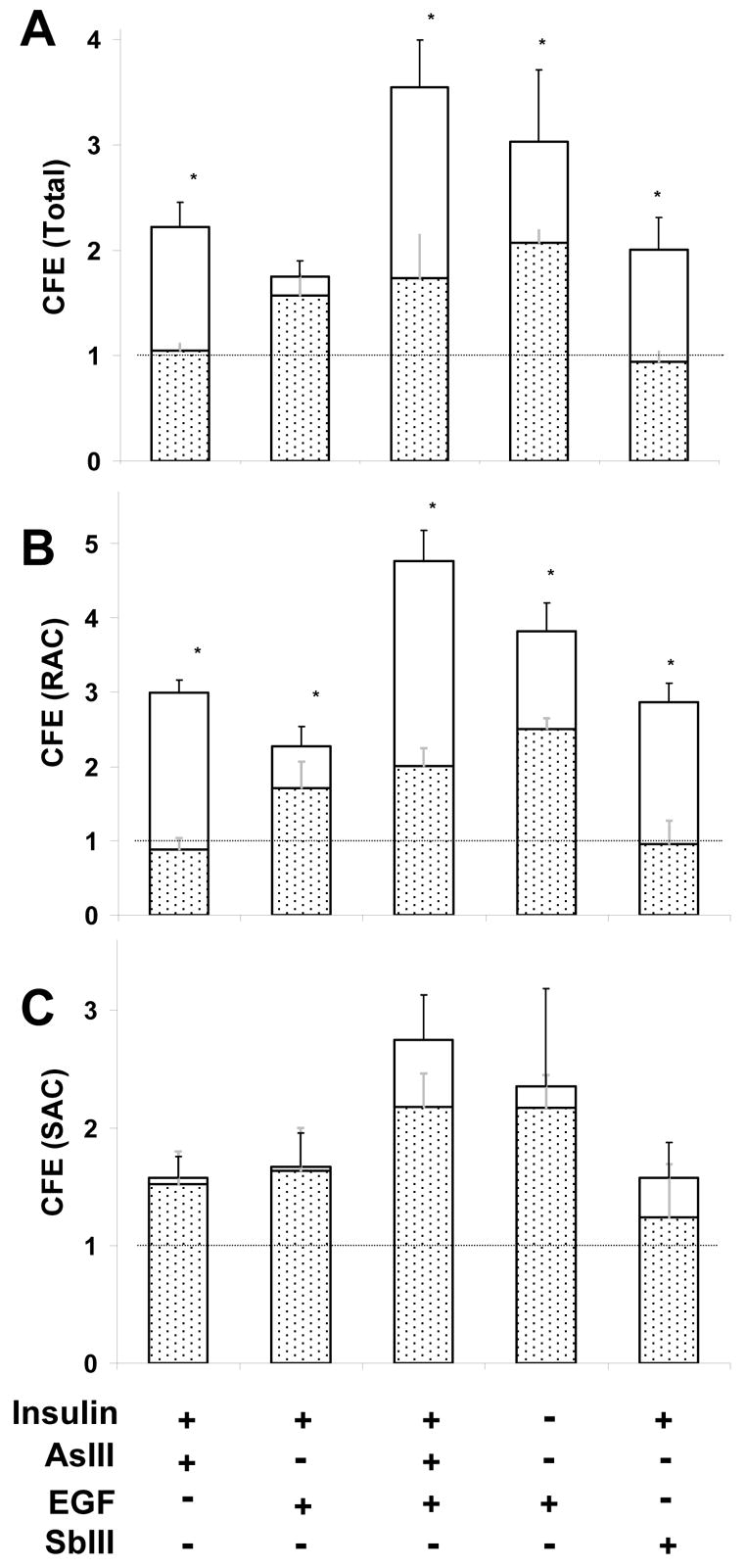
Effect of dominant negative β-catenin on proliferative potential of treated SIK. Cultures were treated as indicated for 3 days starting at 90% confluence before measuring CFE in (A) unfractionated, (B) rapidly adhering cell (RAC) and (C) slowly adhering cell (SAC) populations. The total bar height shows the response of cultures retrovirally infected with vector only (pBabe), and the stippled region shows the lower response of cultures infected with the T3 dominant negative β-catenin construct. Values are relative to untreated cultures in medium with insulin, set at 1. Significant differences (*) between pBabe and T3 infected SIK for indicated treatment are p<0.01 (Student’s t-test with Bonferroni correction) for three experiments in duplicate.
β-Catenin dependent gene transcription
In various cell types, transcription of MMP7 (Brabletz et al, 1999) and PPARδ (He et al, 1999) is increased and BMP-4 is decreased (Baker et al, 1999) by β-catenin. To demonstrate that increased levels of active β-catenin in the keratinocyte cultures resulted in transcriptional changes, expression of these genes was measured by real-time PCR after various treatments. Relative to untreated cultures in standard growth medium, MMP7 and PPARδ expression were both considerably higher in cells treated with arsenite, antimonite or medium lacking insulin (Fig. 7A,B), while BMP-4 gene expression was markedly reduced under these conditions (Fig. 7C). Treatment with EGF led to a small increase (≈50%) for both MMP7 and PPARδ (Fig. 7A,B), but did not much alter BMP-4 expression. Arsenite and EGF co-treatment, which induced the largest increase in active β-catenin levels and CFE, also resulted in the highest expression of MMP-7 and PPARδ (Fig. 7A,B) and the largest suppression of BMP-4 (Fig. 7C). Addition of AG1478 prevented these changes in gene expression, as did expression of the T3 dominant negative β-catenin construct (Fig. 7A,B,C).
Discussion
Modeling of arsenic-induced cancer in vivo has proven difficult, but recent successes have provided much needed animal models (Waalkes et al, 2004). Most relevant to the epidermis, the co-carcinogenic action of arsenic with UV light in hairless mice (Burns et al, 2004) and co-promotion with phorbol ester in Tg.AC mice (Germolec et al, 1998) suggests it acts epigenetically. The present finding that arsenic magnifies and prolongs EGFR action is consistent with this suggestion. Studies in EGFR-deficient mice show that this receptor is essential to maintain the basal cells in a proliferative state so as to support v-rasHa-mediated tumorigenesis (Hansen et al, 2000), and blocking its action prevents UV light-induced tumorigenesis in Tg.AC mice (El-Abaseri et al, 2005). Moreover, overexpressed TGFα in mouse skin acts synergistically with phorbol ester tumor promoter to give epidermal hyperproliferation (Vassar and Fuchs, 1992). Since arsenic greatly enhances UV light-induced epidermal hyperplasia in mice (Burns et al, 2004), present findings suggest that arsenic acts as a co-carcinogen by increasing the pool of epidermal target cells, those with stem cell properties in which carcinomas likely originate (Sell, 2004).
Previous work using high arsenic concentrations for short periods in culture has demonstrated phosphorylation of the EGFR and stimulation of the mitogen activated protein kinase pathway in rat PC12 cells (Chen et al, 1998), a human bronchial epithelial cell line (Wu et al, 1999), a human uroepithelial cell line (Simeonova et al, 2002), human umbilical vein endothelial cells (Nuntharatanapong et al, 2005), A431 epidermoid carcinoma (Huang et al, 2006) and normal human epidermal keratinocytes (Tanaka-Kagawa et al, 2003). This phenomenon, present in many cell types, could affect keratinocyte responses to UV light (Mudipalli et al, 2005). Careful concentration dependence studies in the HaCaT keratinocyte line have shown that lower arsenic levels over longer time periods can produce persistent downstream effects of low magnitude that (as in the other studies) are dependent upon EGFR signaling (Cooper et al, 2004). In contrast to earlier studies of short duration, however, present work at concentrations closer to actual human exposure levels has not detected much extracellular responsive kinase phosphorylation with long term arsenite treatment (unpublished) despite persistent Ras activation. This phenomenon may arise from ERK inactivation by glutathionylation under oxidant conditions (Cross and Templeton, 2004), a well known result of arsenite treatment, with possible assistance from negative feedback as a consequence of Raf phosphorylation (Dougherty et al, 2005).
The mechanism of EGFR activation and stabilization remain to be elucidated. Present experiments performed at low arsenite concentrations have not revealed an influence of src family kinase inhibitors, unlike several earlier studies at higher concentrations (Cooper et al, 2004; Liu and Huang, 2006; Simeonova et al, 2002). A plausible signaling pathway might involve the reversible oxidation of protein tyrosine phosphatases, a well known feature of oxidant-regulated signaling (Meng et al, 2002). The EGFR can interact with several of these phosphatases, among which SHP-1 and SHP-2 are prominent, but their roles in inhibiting or stimulating signaling are complicated (Wang et al, 2006). The recent finding that UV light activates the EGFR through inactivation of protein phosphatase κ (Xu et al, 2006) might also occur with arsenite treatment. Alternatively, oxidative stress elicited by arsenite could interfere with EGFR recycling, as observed from hydrogen peroxide prevention of its ubiquitination in human lung epithelial cells (Ravid et al, 2002). Arsenite could interfere with other aspects of receptor processing such as neddylation (Oved et al, 2006) or caveolin-1-mediated degradation as observed with peroxide exposure (Khan et al, 2006). Arsenite has been reported to cause accumulation of ubiquitinated proteins (Bredfeldt et al, 2004), which could also result in persistent signaling.
Novel aspects of the present findings include the suppressive influence of insulin on proliferative potential after confluence and its antagonism by arsenite treatment. Insulin, originally added to promote cell growth in the absence of serum (Wu and Sato, 1978), is commonly present at generous concentration (5 μg/ml) in culture medium. It is effective for keratinocytes in sparse culture (Tsao et al, 1982), but evidently it acts after confluence to stimulate differentiation and loss from the germinative pool. The relative importance of the insulin receptor and the IGF-1 receptor in mediating these effects is not yet clear (experiments in progress), but in any case the cellular response after confluence could reflect an altered state of signal transducers, including phosphorylation of insulin receptor substrate-1 or insulin receptor substrate-2 (Gual et al, 2005; Sadagurski et al, 2006). Arsenite could plausibly influence protein phosphorylation and in this way simultaneously preserve growth potential and suppress differentiation. Insulin appears to cause degradation of the EGFR in keratinocytes, an effect that arsenite opposes. Cross-talk of the IGF-1R with the EGFR can involve EGFR activation by proteolytic generation of a ligand from the cell surface, such as heparin binding EGF in human embryonic kidney cells (El-Shewy et al, 2004). Preliminary results in present cultures indicate blocking EGF binding to the EGFR does not prevent the increase in CFE observed when insulin is removed. Alternatively, formation of a protein complex at the plasma membrane, demonstrated to occur in human mammary epithelial cells (Ahmad et al, 2004), may result in the observed EGFR down regulation and loss. Present results could be explained by arsenite prevention of EGFR degradation (as shown) and stabilization in an active state, similar to the altered receptor trafficking that occurs with peroxide (Khan et al, 2006). Localization of the EGFR in arsenite-treated cells may help elucidate the mechanism of this hypothetical alteration of processing.
Arsenic treatment clearly was effective in maintaining higher levels of β-catenin, thereby promoting its transcriptional activity. Although proliferation of interfollicular epidermal keratinocytes in mouse skin does not depend upon β-catenin (Posthaus et al, 2002), elevation of this protein reportedly contributes to the high proliferative potential of clonogenic cells (Zhu and Watt, 1999). From the perspective of models proposed for epidermal keratinocyte self renewal (Jones and Watt, 1993), arsenic appears to delay the transition of stem cells to transit amplifying cells. Moreover, overexpression of this protein in MDCK cells increased their cycling after confluence and their survival in suspension culture (Oxford et al, 1999), two features evident in arsenic-treated keratinocytes (Patterson et al, 2005). A variety of tumor types have been found to express β-catenin with stabilizing mutations (Giles et al, 2003), including human pilomatricomas (Chan et al, 1999). Elevated levels of β-catenin, participating in tumor development, have also been attributed to overexpression of EGFR (Lu and Hunter, 2004), overexpression of the transcription factor Np63 in squamous cell carcinomas or loss of p53 (Patturajan et al, 2002). The finding that β-catenin is critical for hair follicle development (Huelsken et al, 2001) raises the intriguing question how much its relatively high expression as a result of arsenic exposure perturbs transcription in that direction despite the limitations of the culture environment. The signaling pathway leading to its elevated levels and transcriptional activity is uncertain, but a recent report that protein tyrosine phosphatase PCP-2 inhibits β-catenin action (Yan et al, 2006) raises the possibility that phosphatase inactivation could contribute to arsenite stabilization of β-catenin.
Immediately beneath arsenic in the periodic table, antimony in the trivalent state shares with arsenite several toxicological properties, including reaction with intracellular thiols, clastogenicity and lack of bacterial mutagenicity (Gebel, 1997). Antimony is a suspect carcinogen, although epidemiological data are confounded by other exposures (e.g., arsenic, lead), and animal experiments have not been definitive (De Boeck et al, 2003). Environmental exposures to antimony generally are low, reflected in trace levels in human urine (Paschal et al, 1998), and contaminated water supplies are much less of a problem than with arsenic, but occupational (McCallum, 2005) and consumer exposures (Shotyk et al, 2006) have been of concern. Use of antimony in treatment of leishmaniasis and recent interest in its potential use in cancer chemotherapy in place of arsenic have stimulated study of its intracellular action, including its interaction with glutathione (Wyllie and Fairlamb, 2006). Previous work has shown that trivalent antimony is nearly as potent as arsenite in suppressing keratinocyte differentiation and inducing heme oxygenase-1, whereas the pentavalent form is inactive (Patterson et al, 2003). The greater efficacy of pentavalent compared to trivalent antimony in treating human leishmaniasis infections evidently reflects facile reduction of SbV to SbIII by the protozoal parasite (Shaked-Mishan et al, 2001) compared to humans. In contrast to arsenic, reduction of pentavalent to trivalent antimony is negligible in human macrophages (Wyllie and Fairlamb, 2006) and keratinocytes (Patterson et al, 2003). On the basis of our findings that antimonite is biologically equivalent in keratinocytes to arsenite, leading to stabilization of the EGFR, elevated β-catenin activity, and preservation of proliferative potential, further study of its carcinogenic or co-carcinogenic action may be warranted. In any case, comparative study of this agent may help elucidate the mechanism of arsenite action.
Fig. 6.
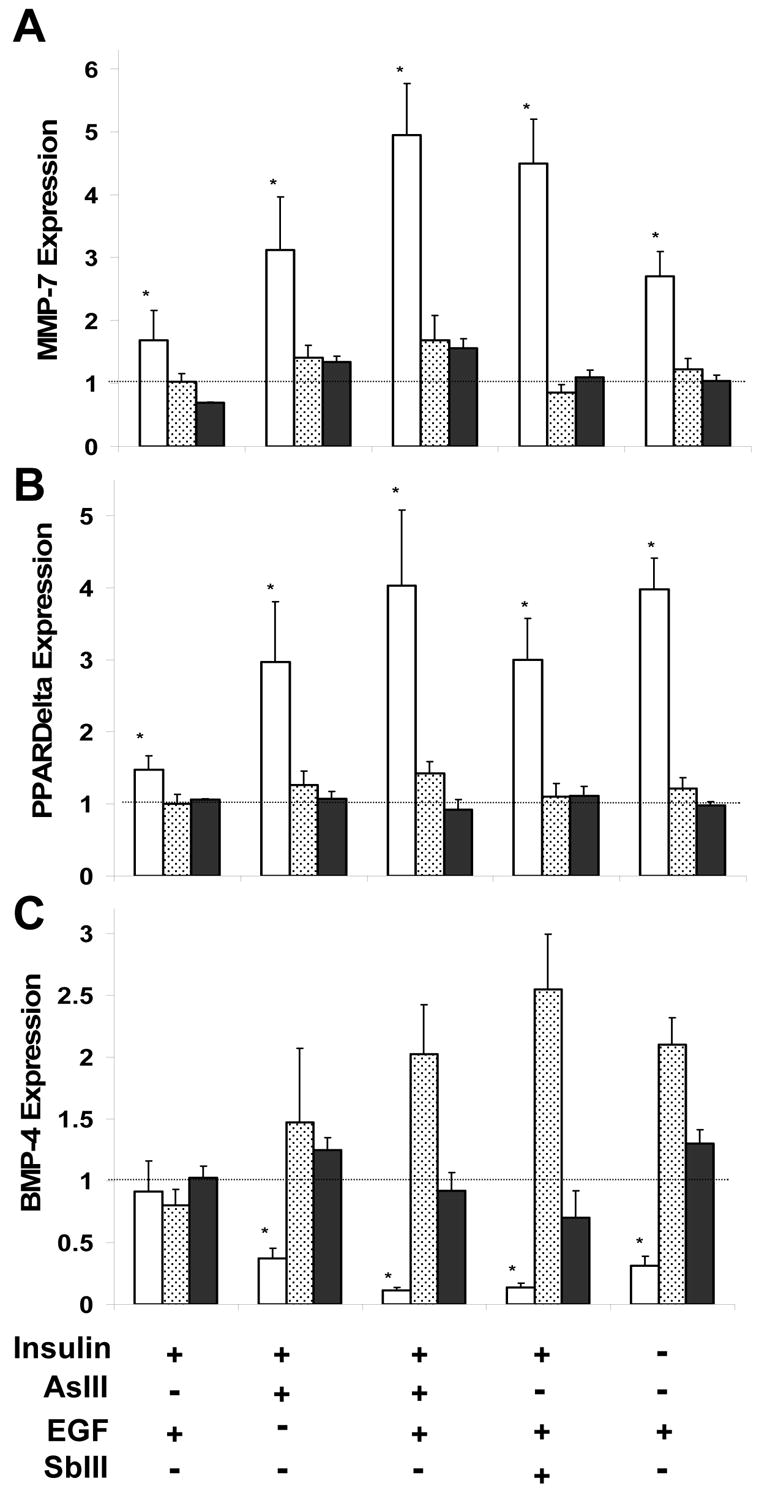
β-Catenin dependent gene expression after arsenite treatment of keratinocytes. 90% confluent cultures of SIK (clear bars), T3-infected SIK (solid bars), and SIK treated with 1 μM AG1478 (stippled bars) were treated for 3 days before measuring expression of (A) MMP7, (B) PPARδ and (C) BMP-4 by real time PCR. AG1478 was added one hr before arsenite, antimonite or EGF. Values are given relative to mRNA in untreated cultures in the presence of insulin, set to 1. Values significantly different (*) from untreated cultures (Student’s t-test with Bonferroni correction) at p<0.01 summarizing four (untreated, arsenite, EGF, arsenite and EGF in SIK) or two (antimonite, no insulin, AG1478, T3) experiments in triplicate.
Acknowledgments
We thank Dr. Marjorie A. Phillips for many helpful suggestions, including exploration of insulin action, and Dr. Barry M. Gumbiner for kindly providing β-catenin T3 cDNA. This research was supported by USPHS grants AR27130, ES07059, ES05707 and ES04699, which had no influence on experimental design or interpretation of the results.
Abbreviations
- BMP-4
bone morphogenetic protein-4
- CFE
colony forming efficiency
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- hEp
human epidermal cells
- IGF-1
insulin-like growth factor-1
- IGF-1R
insulin-like growth factor-1 receptor
- MMP-7
matrix metalloprotease-7
- PCR
polymerase chain reaction
- PPARδ
peroxisome proliferator associated receptor δ
- RAC
rapidly adhering cell
- SAC
slowly adhering cell
- SIK
spontaneously immortalized keratinocytes
Footnotes
Conflict of Interest Statement
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams TE, McKern NM, Ward CW. Signalling by the type 1 insulin-like growth factor receptor: interplay with the epidermal growth factor receptor. Growth Factors. 2004;22:89–95. doi: 10.1080/08977190410001700998. [DOI] [PubMed] [Google Scholar]
- Ahmad T, Farnie G, Bundred NJ, Anderson NG. The mitogenic action of insulin-like growth factor I in normal human mammary epithelial cells requires the epidermal growth factor receptor tyrosine kinase. J Biol Chem. 2004;279:1713–1719. doi: 10.1074/jbc.M306156200. [DOI] [PubMed] [Google Scholar]
- Allen-Hoffmann BL, Rheinwald JG. Polycyclic aromatic hydrocarbon mutagenesis of human epidermal keratinocytes in culture. Proc Natl Acad Sci USA. 1984;81:7802–7806. doi: 10.1073/pnas.81.24.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JC, Beddington RSP, Harland RM. Wnt signaling in Xenopus embryos inhibits Bmp4 expression and activates neural development. Genes Develop. 1999;13:3149–3159. doi: 10.1101/gad.13.23.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrandon Y, Green H. Cell migration is essential for sustained growth of keratinocyte colonies: The roles of transforming growth factor-α and epidermal growth factor. Cell. 1987;50:1131–1137. doi: 10.1016/0092-8674(87)90179-6. [DOI] [PubMed] [Google Scholar]
- Barrandon Y, Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci USA. 1987;84:2302–2306. doi: 10.1073/pnas.84.8.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Dag S, Hlubek F, Kirchner T. β-Catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol. 1999;155:1033–1038. doi: 10.1016/s0002-9440(10)65204-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredfeldt TG, Kopplin MJ, Gandolfi AJ. Effects of arsenite on UROtsa cells: Low-level arsenite causes accumulation of ubiquitinated proteins that is enhanced by reduction in cellular glutathione levels. Toxicol Appl Pharmacol. 2004;198:412–418. doi: 10.1016/j.taap.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Burns FJ, Uddin AN, Wu F, Nadas A, Rossman TG. Arsenic-induced enhancement of ultraviolet radiation carcinogenesis in mouse skin: A dose-response study. Environ Hlth Perspect. 2004;112:599–603. doi: 10.1289/ehp.6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EF, Gat U, McNiff JM, Fuchs E. A common human skin tumour is caused by activating mutations in beta-catenin. Nat Genet. 1999;21:410–413. doi: 10.1038/7747. [DOI] [PubMed] [Google Scholar]
- Chen W, Martindale JL, Holbrook NJ, Liu Y. Tumor promoter arsenite activates extracellular signal-regulated kinase through a signaling pathway mediated by epidermal growth factor receptor. Molec Cell Biol. 1998;18:5178–5188. doi: 10.1128/mcb.18.9.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Graziano JH, Parvez F, Hussain I, Momotaj H, van Geen A, Howe GR, Ahsan H. Modification of risk of arsenic-induced skin lesions by sunlight exposure, smoking, and occupational exposures in Bangladesh. Epidemiol. 2006;17:459–467. doi: 10.1097/01.ede.0000220554.50837.7f. [DOI] [PubMed] [Google Scholar]
- Cooper KL, Myers TA, Rosenberg M, Chavez M, Hudson LG. Roles of mitogen activated protein kinases and EGF receptor in arsenite-stimulated matrix metalloproteinase-9 production. Toxicol Appl Pharmacol. 2004;200:177–185. doi: 10.1016/j.taap.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Cross JV, Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J. 2004;381:675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boeck M, Kirsch-Volders M, Lison D. Cobalt and antimony: Genotoxicity and carcinogenicity. Mutation Res. 2003;533:135–152. doi: 10.1016/j.mrfmmm.2003.07.012. [DOI] [PubMed] [Google Scholar]
- DiGiovanni J, Bol DK, Wilker E, Beltran L, Carbajal S, Moats S, Ramirez A, Jorcano JL, Kiguchi K. Constitutive expression of insulin-like growth factor-1 in epidermal basal cells of transgenic mice leads to spontaneous tumor promotion. Cancer Res. 2000;60:1561–1570. [PubMed] [Google Scholar]
- Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Molec Cell. 2005;17:215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- El-Abaseri TB, Fuhrman J, Trempus CS, Shendrik I, Tennant RW, Hansen LA. Chemoprevention of UV light-induced skin tumorigenesis by inhibition of the epidermal growth factor receptor. Cancer Res. 2005;65:3958–3965. doi: 10.1158/0008-5472.CAN-04-2204. [DOI] [PubMed] [Google Scholar]
- El-Shewy HM, Kelly FL, Barki-Harrington L, Luttrell LM. Ectodomain shedding-dependent transactivation of epidermal growth factor receptors in response to insulin-like growth factor type I. Molec Endocrinol. 2004;18:2727–2739. doi: 10.1210/me.2004-0174. [DOI] [PubMed] [Google Scholar]
- Funayama N, Fagotto F, McCrea P, Gumbiner BM. Embryonic axis induction by the armadillo repeat domain of β-catenin: Evidence for intracellular signaling. J Cell Biol. 1995;128:959–968. doi: 10.1083/jcb.128.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebel T. Arsenic and antimony: Comparative approach on mechanistic toxicology. Chem-Biol Interact. 1997;107:131–144. doi: 10.1016/s0009-2797(97)00087-2. [DOI] [PubMed] [Google Scholar]
- Germolec DR, Spalding J, Yu HS, Chen GS, Simeonova PP, Humble MC, Bruccoleri A, Boorman GA, Foley JF, Yoshida T, Luster MI. Arsenic enhancement of skin neoplasia by chronic stimulation of growth factors. Am J Pathol. 1998;153:737–745. doi: 10.1016/S0002-9440(10)65692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- Green H. The keratinocyte as differentiated cell type. Harvey Lect. 1979;74:101–138. [PubMed] [Google Scholar]
- Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Guo HR, Yu HS, Hu H, Monson RR. Arsenic in drinking water and skin cancers: cell-type specificity (Taiwan, ROC) Cancer Causes Control. 2001;12:909–916. doi: 10.1023/a:1013712203455. [DOI] [PubMed] [Google Scholar]
- Hansen LA, Woodson RL, Holbus S, Strain K, Lo YC, Yuspa SH. The epidermal growth factor receptor is required to maintain the proliferative population in the basal compartment of epidermal tumors. Cancer Res. 2000;60:3328–3332. [PubMed] [Google Scholar]
- He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–345. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hei TK, Filipic M. Role of oxidative damage in the genotoxicity of arsenic. Free Radical Biol Med. 2004;37:574–581. doi: 10.1016/j.freeradbiomed.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Huang HS, Liu ZM, Ding L, Chang WC, Hsu PY, Wang SH, Chi CC, Chuang CH. Opposite effect of ERK1/2 and JNK on p53-independent p21WAF1/CIP1 activation involved in the arsenic trioxide-induced human epidermoid carcinoma A431 cellular cytotoxicity. J Biomed Sci. 2006;13:113–125. doi: 10.1007/s11373-005-9040-z. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. β-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- Jones PH, Watt FM. Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell. 1993;73:713–724. doi: 10.1016/0092-8674(93)90251-k. [DOI] [PubMed] [Google Scholar]
- Khan EM, Heidinger JM, Levy M, Lisanti MP, Ravid T, Goldkorn T. Epidermal growth factor receptor exposed to oxidative stress undergoes Src- and caveolin-1-dependent perinuclear trafficking. J Biol Chem. 2006;281:14486–14493. doi: 10.1074/jbc.M509332200. [DOI] [PubMed] [Google Scholar]
- Kitchin KT. Recent advances in arsenic carcinogenesis: Modes of action, animal model systems and methylated arsenic metabolites. Toxicol Sci. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- Knowlden JM, Hutcheson IR, Barrow D, Gee JM, Nicholson RI. Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: A supporting role to the epidermal growth factor receptor. Endocrinol. 2005;146:4609–4618. doi: 10.1210/en.2005-0247. [DOI] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growthfactor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Liu ZM, Huang HS. As2O3-induced c-Src/EGFR/ERK signaling is via Sp1 binding sites to stimulate p21WAF/CIP1 expression in human epidermoid carcinoma A431 cells. Cell Signaling. 2006;18:244–255. doi: 10.1016/j.cellsig.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Lu Z, Hunter T. Wnt-independent β-catenin transactivation in tumor development. Cell Cycle. 2004;3:571–573. [PubMed] [Google Scholar]
- McCallum RI. Occupational expsoure to antimony compounds. J Environ Monit. 2005;7:1245–1250. doi: 10.1039/b509118g. [DOI] [PubMed] [Google Scholar]
- Meng TC, Fukuda T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Molec Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- Mudipalli A, Owen RD, Preston RJ. The effect of arsenicals on ultraviolet-radiation-induced growth arrest and related signaling events in human keratinocytes. Int J Oncol. 2005;27:769–778. [PubMed] [Google Scholar]
- Nuntharatanapong N, Chen K, Sinhaseni P, Keaney JFJ. EGF receptor-dependent JNK activation is involved in arsenite-induced p21Cip/Waf1 upregulation and endothelial apoptosis. Am J Physiol Heart Circulat Physiol. 2005;289:H99–H107. doi: 10.1152/ajpheart.00901.2004. [DOI] [PubMed] [Google Scholar]
- Oved S, Mosesson Y, Zwanf Y, Santonico E, Shtiegman K, Marmor MD, Kochupurakkal BS, Katz M, Lavi S, Cesareni G, Yarden Y. Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. J Biol Chem. 2006;281:21640–21651. doi: 10.1074/jbc.M513034200. [DOI] [PubMed] [Google Scholar]
- Oxford K, Orford CC, Byers SW. Exogenous expression of β-catenin regulates contact inhibition, anchorage-independent growth, anoikis and radiation-induced cell cycle arrest. J Cell Biol. 1999;146:855–867. doi: 10.1083/jcb.146.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal DC, Ting BG, Morrow JC, Prirkle JL, Jackson RJ, Sampson EJ, Miller DT, Caldwell KL. Trace metals in urine of United States residents: Reference range concentrations. Environ Res, sec A. 1998;76:53–59. doi: 10.1006/enrs.1997.3793. [DOI] [PubMed] [Google Scholar]
- Patterson TJ, Ngo M, Aronov PA, Reznikova TV, Green PG, Rice RH. Biological activity of inorganic arsenic and antimony reflects oxidation state in cultured keratinocytes. Chem Res Toxicol. 2003;16:1624–1631. doi: 10.1021/tx034146y. [DOI] [PubMed] [Google Scholar]
- Patterson TJ, Reznikova TV, Phillips MA, Rice RH. Arsenite maintains germinative state in cultured human epidermal cells. Toxicol Appl Pharmacol. 2005;207:69–77. doi: 10.1016/j.taap.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K, Zangen R, Pollak N, Califano J, Trink B, Ratovitski E, Sidransky D. DeltaNp63 induces β-catenin nuclear accumulation and signalling. Cancer Cell. 2002;1:369–379. doi: 10.1016/s1535-6108(02)00057-0. [DOI] [PubMed] [Google Scholar]
- Posthaus H, Williamson L, Baumann D, Kemier R, Caldelari R, Suter MM, Schwarz H, Muller E. β-Catenin is not required for proliferation and differentiation of epidermal mouse keratinocytes. J Cell Sci. 2002;115:4587–4595. doi: 10.1242/jcs.00141. [DOI] [PubMed] [Google Scholar]
- Ravid T, Sweeney C, Gee P, Carraway KLI, Goldkorn T. Epidermal growth factor receptor activation under oxidative stress fails to promote c-Cbl mediated down-regulation. J Biol Chem. 2002;277:31214–31219. doi: 10.1074/jbc.M204677200. [DOI] [PubMed] [Google Scholar]
- Rheinwald JG, Green H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell. 1975;6:331–344. doi: 10.1016/s0092-8674(75)80001-8. [DOI] [PubMed] [Google Scholar]
- Rheinwald JG, Green H. Epidermal growth factor and the multiplication of human epidermal keratinocytes. Nature. 1977;265:421–424. doi: 10.1038/265421a0. [DOI] [PubMed] [Google Scholar]
- Rice RH, Steinmann KE, deGraffenried LA, Qin Q, Taylor N, Schlegel R. Elevation of cell cycle control proteins during spontaneous immortalization of human keratinocytes. Molec Biol Cell. 1993;4:185–194. doi: 10.1091/mbc.4.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagurski M, Yakar S, Weingarten G, Holzenberger M, Rhodes CJ, Breitkreutz D, LeRoith D, Wertheimer E. Insulin-like growth factor 1 receptor signaling regulates skin development and inhibits skin keratinocyte differentiation. Molec Cell Biol. 2006;26:2675–2687. doi: 10.1128/MCB.26.7.2675-2687.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sell S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol/Hematol. 2004;51:1–28. doi: 10.1016/j.critrevonc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Shaked-Mishan P, Ulrich N, Ephros M, Zilberstein D. Novel intracellular SbV reducing activity correlates with antimony susceptibility in Leishmania donovani. J Biol Chem. 2001;276:3971–3976. doi: 10.1074/jbc.M005423200. [DOI] [PubMed] [Google Scholar]
- Shen S, Alt A, Wertheimer E, Gartsbein M, Kuroki T, Ohba M, Braiman L, Sampson SR, Tennenbaum T. A divergence point in the signaling of insulin and IGF-1-induced proliferation of skin keratinocytes. Diabetes. 2001;50:255–264. doi: 10.2337/diabetes.50.2.255. [DOI] [PubMed] [Google Scholar]
- Shibata MA, Ward JM, Green JE, Merlino G. Enhanced sensitivity to tumor growth and development in multistage skin carcinogenesis by transforming growth factor-α-induced epidermal growth factor receptor activation but not p53 inactivation. Molec Carcinogen. 1997;18:160–170. doi: 10.1002/(sici)1098-2744(199703)18:3<160::aid-mc5>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Shotyk W, Krachler M, Chen B. Contamination of Canadian and European bottled waters with antimony from PET containers. J Environ Monit. 2006;8:288–292. doi: 10.1039/b517844b. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Wang S, Hulderman T, Luster MI. c-Src-dependent activation of the epidermal growth factor receptor and mitogen-activated protein kinase pathway by arsenic. Role in carcinogenesis. J Biol Chem. 2002;277:2945–2950. doi: 10.1074/jbc.M109136200. [DOI] [PubMed] [Google Scholar]
- Sweeney C, Carraway KLI. Negative regulation of ErbB family receptor tyrosine kinases. Brit J Cancer. 2004;90:289–293. doi: 10.1038/sj.bjc.6601500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Kagawa T, Hanioka N, Yoshida H, Jinno H, Ando M. Arsenite and arsenate activate extracellular signal-regulated kinases 1/2 by an epidermal growth factor receptor-mediated pathway in normal human keratinocytes. Br J Dermatol. 2003;149:1116–1127. doi: 10.1111/j.1365-2133.2003.05704.x. [DOI] [PubMed] [Google Scholar]
- Tsao MC, Walthall BJ, Ham RG. Clonal growth of normal human epidermal keratinocytes in a defined medium. J Cell Physiol. 1982;110:219–229. doi: 10.1002/jcp.1041100217. [DOI] [PubMed] [Google Scholar]
- Vassar R, Fuchs E. Transgenic overexpression of transforming growth factor alpha bypasses the need for c-Ha-ras mutations in mouse skin tumorigenesis. Molec Biol Cell. 1992;12:4643–4653. doi: 10.1128/mcb.12.10.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret WJ, Chen Y, Ahmed A, Islam T, Parvez F, Kibriya MG, Graziano JH, Ahsan H. A randomized, double blind placebo-controlled trial evaluating the effects of vitamin E and selenium on arsenic-induced skin lesions in bangladesh. J Occup Environ Med. 2005;47:1026–1035. doi: 10.1097/01.jom.0000183095.45050.97. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Ward JM, Diwan BA. Animal models for arsenic carcinogenesis: Inorganic arsenic is a transplacental carcinogen in mice. Toxicol Appl Pharmacol. 2004;198:377–384. doi: 10.1016/j.taap.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Walton FS, Harmon AW, Paul DS, Drobna Z, Patel YM, Styblo M. Inhibition of insulin-dependent glucose uptake by trivalent arsenicals: Possible mechanism of arsenic-induced diabetes. Toxicol Appl Pharmacol. 2004;198:424–433. doi: 10.1016/j.taap.2003.10.026. [DOI] [PubMed] [Google Scholar]
- Wang N, Li Z, Ding R, Frank GD, Senbonmatsu T, Landon EJ, Inagami T, Zhao ZJ. Antagonism or synergism: The role of tyrosine phosphatases Shp-1 and Shp-2 in growth factor signaling. J Biol Chem. 2006;281:21878–21883. doi: 10.1074/jbc.M605018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Greenhalgh DA, Eckhardt JN, Rothnagel JA, Roop DR. Epidermal expression of transforming growth factor-α in transgenic mice: Induction of spontaneous and 12-O-tetradecanoylphorbol-13-acetate-induced papillomas via a mechanism independent of Ha-ras activation or overexpression. Molec Carcinogen. 1994;10:15–22. doi: 10.1002/mc.2940100104. [DOI] [PubMed] [Google Scholar]
- Wertheimer E, Spravchikov N, Trebicz M, Gartsbein M, Accili D, Avinoah I, Nofeh-Moses S, Sizyakov G, Tennenbaum T. The regulation of skin proliferation and differentiation in the IR null mouse: Implications for skin complications of diabetes. Endocrinol. 2001;142:1234–1241. doi: 10.1210/endo.142.3.7988. [DOI] [PubMed] [Google Scholar]
- Wu R, Sato GH. Replacement of serum in cell culture by hormones: A study of hormonal regulation of cell growth and specific gene expression. Exp Cell Res. 1978;4:427–448. doi: 10.1080/15287397809529669. [DOI] [PubMed] [Google Scholar]
- Wu W, Graves LM, Jaspers I, Devlin RB, Reed W, Samet JM. Activation of the EGF receptor signaling pathway in human airway epithelial cells exposed to metals. Am J Physiol. 1999;277:L924–L931. doi: 10.1152/ajplung.1999.277.5.L924. [DOI] [PubMed] [Google Scholar]
- Wyllie S, Fairlamb AH. Differential toxicity of anitmonial compounds and their effects on glutathione homeostasis in a human leukemia monocyte cell line. Biochem Pharmacol. 2006;71:257–267. doi: 10.1016/j.bcp.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Xu Y, Shao Y, Voorhees JJ, Fisher GJ. Oxidative inhibition of receptor-type protein-tyrosine phosphatase κ by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes. J Biol Chem. 2006;281:27389–27397. doi: 10.1074/jbc.M602355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HX, Yang W, Zhang R, Chen L, Tang L, Zhai B, Liu SQ, Cao HF, Man XB, Wu HP, Wu MC, Wang HY. Protein-tyrosine phosphatase PCP-2 inhibits β-catenin signaling and increases E-cadherin-dependent cell adhesion. J Biol Chem. 2006;281:15423–15433. doi: 10.1074/jbc.M602607200. [DOI] [PubMed] [Google Scholar]
- Zhu AJ, Watt FM. β-Catenin signaling modulates proliferative potential of human epidermal keratinocytes independently of intercellular adhesion. Development. 1999;126:2285–2298. doi: 10.1242/dev.126.10.2285. [DOI] [PubMed] [Google Scholar]