

Abstract
To efficiently generate varicella-zoster virus (VZV) mutants, we inserted a bacterial artificial chromosome (BAC) vector in the pOka genome. We showed that the recombinant VZV (VZVBAC) strain was produced efficiently from the BAC DNA and behaved indistinguishably from wild-type virus. Moreover, VZV's cell-associated nature makes characterizing VZV mutant growth kinetics difficult, especially when attempts are made to monitor viral replication in vivo. To overcome this problem, we then created a VZV strain carrying the luciferase gene (VZVLuc). This virus grew like the wild-type virus, and the resulting luciferase activity could be quantified both in vitro and in vivo. Using PCR-based mutagenesis, open reading frames (ORF) 0 to 4 were individually deleted from VZVLuc genomes. The deletion mutant viruses appeared after transfection into MeWo cells, except for ORF4, which was essential. Growth curve analysis using MeWo cells and SCID-hu mice indicated that ORF1, ORF2, and ORF3 were dispensable for VZV replication both in vitro and in vivo. Interestingly, the ORF0 deletion virus showed severely retarded growth both in vitro and in vivo. The growth defects of the ORF0 and ORF4 mutants could be fully rescued by introducing wild-type copies of these genes back into their native genome loci. This work has validated and justified the use of the novel luciferase VZV BAC system to efficiently generate recombinant VZV variants and ease subsequent viral growth kinetic analysis both in vitro and in vivo.
Varicella-zoster virus (VZV) is a human alphaherpesvirus that is a significant pathogen in the United States. Primary infection of VZV typically occurs during childhood and leads to varicella (chickenpox). Like all herpesviruses, VZV establishes lifelong latency in the host, specifically in trigeminal ganglia and dorsal root ganglia. The end sequela of VZV reactivation is herpes zoster (shingles), which is characterized by a painful vesicular rash of dermatomes that can lead to chronic postherpetic neuralgia (1, 10).
Beginning in 1995, the U.S. government recommended vaccination of all children with the attenuated Japanese Oka varicella virus (vOka) (28). This has significantly improved prophylaxis against varicella infection. The effectiveness of the vaccination in children is estimated to be between 72% and 98% (11). However, breakthrough rates of varicella have been calculated to be between 2% and 34% (11). As a result of the vaccine recommendation, there is speculation that a rise in herpes zoster in older adults may occur because of less exposure to wild-type VZV to boost natural immunity (30).
Despite the success in preventing and controlling VZV infection, many aspects of its pathogenesis are not completely understood. VZV possesses the smallest genome among all human herpesviruses; it contains a DNA genome of 125 kb that bears 70 unique open reading frames (ORFs). Although many of its ORF products have amino acid sequences homologous to those of herpes simplex virus type 1, less than 20% of the VZV genome has been functionally characterized. This lack of knowledge is partly due to the difficulty of generating mutants to investigate gene function and the long period of time needed to establish pure clones using conventional techniques with mammalian cells. A more detailed understanding of the molecular biology, replication, and pathogenesis of VZV will aid in discovering optimal prophylactic compounds and vaccine strains and in improving therapeutic regimens for varicella infection and zoster.
In recent years, a major advance in VZV genetics was the cloning of overlapping segments of the VZV genome into four cosmids (6, 21). This system allows the creation of recombinant VZV by cotransfection of four overlapping cosmids, one of which can contain a mutation in a desired ORF, representing the entire genome of the parental Oka (pOKA) strain. An attractive alternative for generating recombinant VZV is the recent development of the VZV bacterial artificial chromosome (VZVBAC) (18). The BAC technology provides easy and efficient manipulation of the viral genome and rapid isolation of these recombinant viruses (2).
Other obstacles in understanding VZV pathogenesis include its narrow host range and the dramatic differences in its replication cycles when studied in vitro versus in vivo (1, 8). For instance, VZV infection is highly cell associated in cell culture. In contrast, as observed with human tissue biopsy samples, VZV propagates by both lytic (release of cell-free virus) and cell-to-cell infection. Thus, merging the observations from in vitro and in vivo studies may be difficult. Various in vitro and in vivo systems have been developed to analyze VZV replication and pathogenesis. In addition to various cell lines (26), intact portions of human organs provide a more relevant environment with which to study VZV pathogenesis, because they allow VZV infection to be studied in fully differentiated human cells in their normal tissue architecture and microenvironment. Development of the model of the severe combined immunodeficient mouse with human tissue xenograft (SCID-hu) greatly facilitates investigation of VZV pathogenesis in vivo. Results from histopathological studies have shown that this tissue graft strategy is an accurate representation of VZV pathogenesis in human tissues (1, 3, 13, 33). Additionally, an ex vivo human skin organ culture for VZV skin tropism was recently developed (29).
In the present study, the full-length pOka genomic DNA was first cloned as a BAC, and then a luciferase gene was inserted into this viral genome. The BAC DNA isolated from Escherichia coli was used to transfect human MeWo cells to generate a luciferase-expressing VZV (VZVLuc) strain. This VZVLuc strain grows normally and can be monitored both in vitro and in vivo by enzyme activity. Using the highly efficient bacterial recombination system, five VZV ORFs, from ORF0 to ORF4, were deleted individually, and their functions for viral replication were analyzed in both in vitro and in vivo systems.
MATERIALS AND METHODS
Cells, cosmids, and viruses.
Human melanoma (MeWo) cells and primary human foreskin (HF) fibroblasts were grown in Dulbecco's minimal essential medium supplemented with 10% fetal calf serum, 100 U of penicillin-streptomycin/ml, and 2.5 μg of amphotericin B/ml as previously described and used to propagate VZV (15, 17) in vitro. Each of the four cosmids, pvFsp73, pvSpe14, pvPme19, and pvSpe23, contains overlapping genomic DNA fragments of VZV derived from the pOka clinical isolate previously described (21). Recombinant pOka virus was derived by cotransfection of these four cosmids (21).
Plasmids.
A BAC vector, pUSF-3, was described in reference 15. pUSF-4 was constructed by removing a ClaI site from the green fluorescent protein (GFP) expression cassette of pUSF-3. pUSF-5 was formed by digesting pUSF-4 with HindIII to remove two human cytomegalovirus (HCMV) fragments and religation of the plasmid. pUSF-6 was constructed from pUSF-5 by inserting two fragments of VZV DNA with added sequences for BamHI, HindIII, and LoxP sites. PCR primers were designed to amplify two fragments of ∼500 bp between ORF60 and ORF61 of the VZV pOka strain at genomic sites 102080 to 102560 and 102564 to 103039 (NCBI nucleotide accession no. AB097933). The four primers are 5′-GGA TCC AAC GTA CCC CAA ACT TAA AAC GCT C and 5′-AAG CTT ATA ACT TCG TAT AGC ATA CAT TAT ACG AAG TTA TAT TAA AAT GGC CGG AAG GTG GCG G; and 5′-AAG CTT ATA ACT TCG TAT AAT GTA TGC TAT ACG AAG TTA TAA CTT TAA TTG CTA TAA GAC ATA CCC AAA C and 5′-GGA TCC TGT TGG GAG GGG GAA GGA AAT CG. The fragments were then cloned separately into pCR4-TOPO (Invitrogen), sequenced to verify the products, digested with BamHI and HindIII, and finally combined in a triple ligation into pUSF-5 at the HindIII site, creating pUSF-6. pGEM-T TA cloning vector was purchased from Promega (Madison, WI). pGEM-lox-zeo was used for rescue virus generation as described in reference 20. pGL3-hyg was derived from plasmid pGL3-control (Promega) by replacing its simian virus 40 (SV40) enhancer sequence with a hygromycin resistance gene expression cassette from pIJ963 (a gift from Issar Smith). pGEM-oriV/kan1 was described in reference 31.
Construction of the VZV BAC clone.
The cosmid pvSpe23 was transfected into E. coli strain DY380 (27, 32). Electrocompetent DY380 cells were prepared after incubating log-phase cells at 42°C for 15 min as described previously (31, 32). pUSF-6 was linearized by digestion with BamHI and transfected into DY380 cells containing pvSpe23, and recombinant clones were selected on LB plates with 12.5 μg/ml chloramphenicol. The resulting BAC vector-containing cosmid DNA, pvSpe23-pUSF6, along with three other cosmids, pvFsp73, pvSpe14, and pvPme19, were linearized by digestion with AscI and mixed in water to final concentrations of 100 ng/μl for pVFsp73, pVSpe14, and pVPme2 and 50 ng/μl for pvSpe23-pUSF6. Transfections were done with MeWo cells by using 30 μl of the cosmid mixture in 31.5 μl of 2 M CaCl2 in water and HEPES-buffered saline (14). Green fluorescent plaques were visualized using fluorescent microscopy. Infected cells were split twice at a ratio of 1:3 to allow the virus to spread through the monolayer. The circular VZV DNA containing the BAC vector was isolated from infected MeWo cells and transfected into E. coli (Gene Hogs, Invitrogen, Carlsbad, CA), and transformants resistant to chloramphenicol were selected as described previously (15). BAC DNA from the resulting colonies was purified from 4-ml cultures, and the full-length VZV genomic DNA in these clones was verified by HindIII digestion and restriction fragment length polymorphism analysis.
Generation of VZVBAC from BAC DNA isolated from E. coli.
E. coli DY380 with VZVBAC was cultured in 500 ml of LB medium with 12.5 μg/ml chloramphenicol at 32°C for 20 h. VZVBAC DNA was isolated using a Nucleobond Maxiprep BAC DNA isolation kit (BD Biosciences, Palo Alto, CA) and used to transfect MeWo cells by using FuGene6 transfection reagent (Roche, Indianapolis, IN) according to the manufacturer's standard protocol. The ratio of DNA to transfection reagent was 1.5 μg:6 μl for a transfection reaction mixture in a well (35 mm) of six-well plates. VZV plaques were normally visualized after 3 days posttransfection. To remove the BAC vector (flanked by two loxP sites) from the VZV genome, a Cre expression vector was cotransfected with VZV BAC DNA as described in reference 15.
Construction of VZVLuc.
To generate VZVLuc, the 100-bp sequence between ORF65 and ORF66 was replaced with a luciferase-hygromycin-resistant gene (Hygr) cassette in which the luciferase gene is driven by an SV40 early promoter by homologous recombination. The luciferase-Hygr expression cassette was amplified from the pGL3-hyg plasmid by PCR using a QIAGEN Hotstar DNA polymerase kit and two primers (5′-GTC AGC TAT ACC GAC ATT CTC ACA AAG AGG TAA AGT TAC CCG AGC TCT TAC GCG TGC TAG and 5′-TGA CAA AAA GGG GAG GGG TTA AAC ATA ACT TAC AAA TAT GTC AGG CGC CGG GGG CGG TGT). This PCR product contained a luciferase-Hygr cassette flanked by the 40-bp homologous sequences targeting the interval sequence between ORF65 and ORF66. PCR product was digested with DpnI (New England Lab) and purified by using a PCR purification kit (QIAGEN). About 200 ng of PCR product was transformed via electroporation at 1.6 kV and 250 μF by using a Bio-Rad Gene Pulser II in E. coli DY380 carrying the VZVBAC. The recombinants were selected on agar plates containing 50 μg/ml hygromycin (Sigma) at 32°C.
Construction of ORF0 to ORF4 deletion mutant viruses.
To delete each ORF, two primers were designed to contain 20 bp of nucleotides homologous to the kanamycin resistance gene (Kanr) cassette and 40 bp of nucleotides homologous to the flanking sequences adjacent to either the start or stop codon of the ORF being targeted for deletion. These primers for deletion include the following sequences: for ORF0, 5′-AAC CCG CGC CTT TTG CGT CCA CCC CTC GTT TAC TGC TCG GGC TCT TGT TGG CTA GTG CGT A and 5′-GCA AGC GAG AAT AAA TAC CTT CCC CTT CCG GAC AGT AGT TTC TGC CAG TGT TAC AAC CAA; for ORF1, 5′-ACT CAA CTA CAT GAA ACT ACT GTC CGG AAG GGG AAG GTA TGC TCT TGT TGG CTA GTG CGT A and 5′-ATA AGC AAA TCC TGT TAA TAT TAT ATT TTT GGG ATC CGC ATC TGC CAG TGT TAC AAC CAA; for ORF2, 5′-TAA TAG CTA TTA TCG TAA CCC ACC CCC GTA AAA TCA TAA AGC TCT TGT TGG CTA GTG CGT A and 5′-AAA TAC GTA CAA TCG AAA AAA GGT GTA TTT TAT TTA GTG ATC TGC CAG TGT TAC AAC CAA; for ORF3, 5′-CTT TTT TCG ATT GTA CGT ATT TTT ATT TAA ATG TGT AGT TGC TCT TGT TGG CTA GTG CGT A and 5′-GGG TAA AAC ACA CAC CAG ACG TGT ACC GAA CGT TTA ATT ATC TGC CAG TGT TAC AAC CAA; and for ORF4, 5′-TTA GTA TGT TTT GAC AAG CAT GAA AAA GGT ATT TTT TAT TGC TCT TGT TGG CTA GTG CGT A and 5′-AGG CAA CTG CAA ACA CGC AAT TGT CAG ATA TTT TGC AGC CTC TGC CAG TGT TAC AAC CAA. The Kanr cassette was amplified from the plasmid pGEM-oriV/kan1. PCR products were digested with DpnI and purified by a gel extraction kit (QIAGEN). About 200 ng of each PCR product was transformed into the DY380 cells carrying the VZVLuc BAC via electroporation under the conditions described above. The recombinants were selected on agar plates containing 30 μg/ml kanamycin (Sigma) at 32°C. VZV mutant DNA was isolated from E. coli and verified by the HindIII digestion patterns and PCR analysis and then were transfected into MeWo cells to produce mutant VZV viruses as described above.
Construction of rescue viruses for ORF0D and ORF4D.
To generate rescue viruses, ORF0 and ORF4 were amplified by PCR using an Invitrogen high-fidelity Platinum Taq DNA polymerase kit and cloned into the plasmid pGEM-lox-zeo between the NotI and BglII sites for ORF0 and between the SpeI and NdeI sites for ORF4 to form pGEM-zeo-ORF0 and pGEM-zeo-ORF4, respectively. Both cloned fragments were verified by sequencing analysis, and no coding errors were introduced into these ORFs by PCR amplification. Then, the zeocin resistance (Zeor)-ORF0 cassette was amplified by PCR from pGEM-zeo-ORF0 using two primers (5′-AAC CCG CGC CTT TTG CGT CCA CCC CTC GTT TAC TGC TCG GAT GGC GAC CGT GCA CTA CTC and 5′-GCA AGC GAG AAT AAA TAC CTT CCC CTT CCG GAC AGT AGT TGG ATG GAT CCA TAA CTT CGT), and the Zeor-ORF4 cassette was amplified by PCR from pGEM-zeo-ORF4, using two other primers (5′-TTA GTA TGT TTT GAC AAG CAT GAA AAA GGT ATT TTT TAT TGG ATG GAT CCA TAA CTT CGT and 5′-AGG CAA CTG CAA ACA CGC AAT TGT CAG ATA TTT TGC AGC CAT GGC CTC TGC TTC AAT TCC AAC). These PCR products contain 40 bp of VZV genomic adjacent sequences homologous to those of the Kanr cassettes that are located in the deleted ORF0D and ORF4D loci. The resultant PCR products were treated with DpnI and transformed into the DY380 cells carrying ORF0D and ORF4D BAC clones via electroporation as described above. The recombinants were selected on agar plates containing 50 μg/ml zeocin (Invitrogen) and 12.5 μg/ml chloramphenicol at 32°C. The correct rescue clones (ORF0R to ORF4R) were first confirmed by their antibiotic sensitivities. The correct clones should be resistant to chloramphenicol, hygromycin, and zeocin and sensitive to kanamycin and ampicillin. These clones were further confirmed by restriction digestion and PCR analysis. Maxiprep DNA was purified from each and used to transfect MeWo cells to produce the rescue viruses as described above.
Titering and growth curve analysis.
Titers for each recombinant virus were determined by infectious focus assay. MeWo cells were seeded in six-well plates and mixed with different dilutions of infected cells. Green fluorescent VZV plaques were counted by using a fluorescent microscope, at 3 days postinfection (dpi). Growth curve analyses were carried out using two different methods. For a conventional way of using the infectious center assay, MeWo cells were mixed with 100 PFU of infected MeWo cells and seeded in six-well plates. At each day of infection, three wells of infected cells were collected, and titers were determined. The numbers of plaques from each day were averaged, and a growth curve was generated. For a growth curve using a bioluminescence assay, six-well plates of MeWo cells were infected with 100 PFU of infected MeWo cells, as described above. After 24 h of infection, d-luciferin was added to the cultured wells to a final concentration of 150 μg/ml. After a 10-min incubation at 37°C, the bioluminescence in different wells was recorded simultaneously using an in vivo imaging system (IVIS; 50 Series; Xenogen Corporation, Alameda, CA) following the manufacturer's instruction. After measurement, the luciferin-containing media were replaced by regular cell culture media. This measurement was repeated every 24 h for 7 days. Bioluminescence signals from manually defined regions of interest were quantified and analyzed by using LivingImage image analysis software (Xenogen).
Infection of human T-cell and skin xenografts and measurement of infection in the SCID-hu mouse model.
Thymus-liver conjoint implants were engrafted in male CB-17 SCID/beige mice. The construction and use of thymus-liver implants were described in detail previously (4, 17). About 3 months after implantation, the thymus-liver implants were surgically exposed and directly injected with 2 × 103 to 4 × 103 PFU of the VZVLuc-infected MeWo or HF cells (10 to 20 μl of cell suspension). VZV replication in vivo was measured using an IVIS (Xenogen). At each day postinfection, 250 μl of luciferase substrate, d-luciferin (15 mg/ml), was administered to mice by intraperitoneal injection, and bioluminescence was observed 10 min later as photon flux (photons/s/cm2/steradian) in the regions of interest above the thymus-liver implants. Human fetal skin xenografts were introduced into SCID mice subcutaneously as full-thickness dermal grafts as described previously (17). Four weeks after implantation, the VZVLuc virus was inoculated, and viral growth was monitored using an IVIS, exactly as described above.
RESULTS
Construction of VZVBAC.
In order to use the full power of molecular genetic techniques to efficiently isolate recombinant VZV, the pOka genome was cloned as a BAC, using a construction approach similar to that used for HCMV BAC (15). The VZVBAC was constructed from a vector, pUSF-6, that contains the prokaryotic replication origin (ori) gene, the replication and partition function (repE, parA, and parB) genes, the chloramphenicol resistance (Camr) gene, and the GFP eukaryotic expression cassette (Fig. 1A). The pUSF-6 vector, flanked by two 500-bp VZV fragments, was inserted between ORF60 and ORF61 in VZV cosmid pvSpe23 (Fig. 1B) by homologous recombination using a highly efficient recombination system (32). Then, the BAC-containing cosmid was cotransfected with three complementary cosmids (21) into MeWo cells to generate a recombinant virus. Green fluorescent plaques were observed a few days posttransfection (Fig. 1C), indicating that the recombinant viruses harboring the BAC sequence were generated. Circular VZV DNA was purified from infected cells and transformed into E. coli. Two days later, many chloramphenicol-resistant colonies were obtained. The VZVBAC DNA was then isolated from E. coli.
FIG. 1.

Construction of VZVBAC. (A) A BAC vector, pUSF-6, contains the prokaryotic replication origin (ori) gene, the replication and partition (repE, parA, and parB) genes, the Camr gene, a green fluorescent protein (gfp) gene, two 500-bp VZV fragments (gray bars a and b), and two loxP sites (two white bars). To insert this BAC vector into a VZV cosmid, pUSF-6 was digested by BamHI, resulting in a linear fragment. (B) Schematic diagram of the VZV pOka genome shows that VZV contains a 125-kb genome with unique long (UL) and unique short (US) segments. Four cosmids containing overlapping VZV genomic segments are shown below the diagram. pUSF-6 was inserted between ORF60 and ORF61 in a VZV cosmid, pvSpe23, by homologous recombination. (C) The pvSpe23 containing a BAC vector was cotransfected with three other VZV cosmids into MeWo cells, and the resulting recombinant virus was replicated and produced a green fluorescent plaque. (D) The growth curve of VZVBAC was compared to that of wild-type pOka. PFU/ml at each dpi were recorded. This result indicates that VZVBAC (green) has no detectable growth defect; it grows as well as the parental virus (red) in vitro.
Generation and analysis of VZVBAC viruses.
The integrity and stability of VZVBAC DNA were first confirmed by restriction enzyme digestion. HindIII digestion of VZVBAC DNA generated the predicted digestion pattern with the sum of ∼130 kb, indicating that no large deletions and rearrangements in VZVBAC DNA were detectable (see Fig. 4B, wild type [WT]). To closely examine whether base-pair changes could be detected in the VZVBAC genome after synthesis in E. coli, the ORF62/71 genes, which encode an immediate-early transactivating protein, were sequenced. The 3.9-kb coding sequences were amplified by PCR, cloned into pGEM-T vector, and sequenced as described previously (23). The ORF62/71 sequences in VZVBAC are identical to those of the published pOka strain (accession no. AB097933). These results suggest that the BAC DNA in E. coli is stable.
FIG. 4.

Analysis of VZV ORF0 to ORF4. (A) Schematic diagram of the genomic organization of VZV ORF0 to ORF5 (1). Each ORF, from 0 to 4, was replaced by a Kanr expression cassette to generate the deletion mutant as indicated for ORF4 (2). To create a rescue virus for the ORF4 deletion, ORF4 was cloned into pGEM-zeo-loxP, and the loxP-zeo-ORF4 cassette was amplified by PCR and used to replace the Kanr cassette by homologous recombination (3). The zeocin-resistant gene (Zeor) can be removed from the viral genome by introducing Cre recombinase (4). (B) Digestion analysis of VZV wild-type and recombinant viral BAC clones. VZV DNA from the wild type (WT), the ORF0 to ORF4 deletion clones (0D to 4D), and the ORF0D and ORF4D rescue clones (0R and 4R) was digested with HindIII and loaded onto a 0.5% agarose gel. In order to clearly show both smaller fragments and large fragments, two photos were taken. The lower panel was taken at an earlier time, and the upper panel was taken at a later time. Sizing markers (MR) are indicated at the left. (C) Growth curve analysis of VZV deletion mutants in vitro. One hundred PFU of each deletion mutant and VZVLuc (WT) were used to infect MeWo cells in six-well dishes in triplicate. Luminescence was measured using IVIS every day for 7 days after d-luciferin was applied to the cultured media. Total photon flux in each well (photons/s/cm2/steradian) was measured, and the values from triplicates were averaged. Growth curves were generated when the averaged photon counts for each day were plotted. (D) Growth curve analysis of VZV deletion mutants in vivo. SCID-hu mice with thymus-liver implants were inoculated with 4 × 103 PFU VZVLuc or VZV deletion mutant strains as indicated. VZV replication was measured by IVIS daily for 1 week as photon flux in the regions of interest above the thymus-liver implants. Each line represents an average of data from 2 or 3 mice. An uninfected SCID-hu thymus-liver mouse was injected with d-luciferin, and the background luminescence was measured (pink line). (E) Growth curve analysis of VZV ORF0 and ORF4 deletion rescue viruses in vitro. The conditions in this assay were same as those described for panel B. (F) Growth curve comparison of measurements by luminescence assay and infectious center assay. MeWo cells were grown in six-well dishes in triplicate. The cells were infected with 100 PFU of wild-type (WT), ORF0 deletion (ORF0D), and ORF0D rescue (ORF0R) viruses, respectively. Growth curve analyses were carried out by luminescence assays (LA) and infectious center assays (IA) as described above. Total photon counts in each well were measured using IVIS, averaged, and plotted on the left. Total plaques per well were counted, averaged, and plotted on the right.
To make VZV from the BAC DNA, three independent VZVBAC DNA clones were transfected into MeWo cells. Many green fluorescent VZV plaques appeared from each clone at about 3 days posttransfection. To examine whether the VZV strains produced from VZVBAC DNA grew like the parental pOka virus, the growth properties of VZVBAC and pOka virus in MeWo cells were compared. The results showed that both viruses grew at a similar rate (Fig. 1D).
The stability of the GFP marker in the viral genome was also tested. MeWo cells were infected with VZVBAC and continuously passed four times (1:10 dilution) over 2 weeks. The plaques were examined using a fluorescent microscope. All VZV-infected cells fluoresced green, suggesting that the GFP marker is stable in the viral genome.
These experiments demonstrated that VZVBAC DNA is infectious, that this method was efficient to produce stable VZV viruses, and that no growth defect was detected in the resulting viruses in vitro.
Generation of VZVLuc.
It is difficult to monitor VZV replication in vivo. This problem can be solved by the addition of the luciferase gene to the VZV genome. Luciferase has been widely used as a reporter gene for transcriptional regulation studies. Its application has expanded to in vivo bioluminescence imaging in studies of bacterial and viral infection (9).
To generate a VZVLuc strain, a firefly luciferase expression cassette was inserted into the intergenic region between ORF65 and ORF66 of the VZVBAC genome. This clone was then transfected into MeWo cells to produce the VZVLuc strain. The properties of VZVLuc were analyzed. Growth curve analysis was first performed and showed that VZV expressing luciferase grows like its parental VZVBAC (data not shown). The luciferase activity from infected cells was then measured. The cells infected by VZVLuc produced high levels of luciferase activity, while the cells infected with VZVBAC did not (Fig. 2A). Bioluminescence from VZVLuc-infected cells was then visualized and measured using IVIS (Xenogen) imaging (22). MeWo cells were grown in six-well culture dishes and infected with either VZVBAC (Fig. 2B, upper panel, two left wells) or VZVLuc (Fig. 2B, upper panel, two right wells). Three days postinfection, many green fluorescent plaques were observed for all wells, using fluorescence microscopy (Fig. 2B, lower panel), indicating that the cells were efficiently infected by both viruses. When the substrate of luciferase, d-luciferin, was added to the culture medium, strong bioluminescence was detected only from VZVLuc-infected cells (Fig. 2B, upper panel, two right wells), not from VZVBAC-infected cells (Fig. 2B, upper panel, two left wells).
FIG. 2.

Generation and analysis of the VZVLuc strain. (A) Luciferase assay. MeWo cells were infected with VZVLuc or VZVBAC for 2 days, and luciferase activity was measured. The cells infected with VZVLuc showed a high level of luciferase activity, while the parental VZVBAC strain had no activity. (B) Bioluminescence measurement. Two wells of a six-well dish of MeWo cells were infected with VZVBAC (upper left), and two were infected with VZVLuc (upper right). Two days postinfection, d-luciferin substrate was added to the cultured wells, and bioluminescence was measured using IVIS imaging. Bioluminescence can be detected only in VZVLuc-infected cells. The intensities were indicated as pseudocolors, as shown by an intensity scale bar at the top; higher intensity is represented by a warmer color, and lower intensity is represented with a cooler color. The infection of these wells was verified by showing GFP-positive plaques (bottom panel). (C) Correlation of luminescence and plaque numbers. Growth curves generated by an infectious center assay (black curve and left scale) and a bioluminescence assay (green curve and right scale) were compared. (D) The growth curve comparison of VZVLuc with and without the BAC vector. MeWo cells were infected with VZV with the BAC vector (green) and without the BAC vector (blue). Their grow curves were generated by bioluminescence assay. (E) Monitoring VZV replication in vivo. SCID mice with human thymus-liver implants were inoculated with 4 × 103 PFU VZV-infected cells. At 7 days postinfection, a luciferase substrate, d-luciferin, was injected, and bioluminescence was observed 10 min later using IVIS imaging and overlaid on the mouse images.
Using bioluminescence to quantify growth kinetics of VZV would significantly reduce the effort and increase the accuracy of the infectious center assay. We investigated whether bioluminescence intensity correlated with viral titers. Growth curves of VZVLuc were compared using the conventional infectious center assay and the bioluminescence assay. For the infectious center assay, 21 wells were used. The contents of three wells were collected each day for 7 days. The titer of each sample was determined to produce a growth curve (Fig. 2C, black curve). For the bioluminescence assay, only three wells of infected cells were used for the entire experiment. The bioluminescence from infected cells was measured each day for 7 days, and a growth curve was generated. After 7 days of infection, the signals started to slowly decline due to deterioration of the cultured cells. As shown in Fig. 2C (green curve), these two curves correlated well, indicating that semiquantifying bioluminescence can be an alternative way to measure viral titers and used for growth curve assays.
To determine whether the BAC vector in the VZV genome has any effect on viral growth, the growth curves of VZVLuc with and without the BAC vector were compared using bioluminescence assay. As shown in Fig. 2D, both viruses replicated equally well, indicating that the BAC vector has no discernible effect on VZV replication in vitro.
Live-image analysis of VZV replication with SCID-hu mice.
Since VZV is restricted to human cells, mouse models for VZV studies are limited to those that engraft human tissues in immunodeficient mice. The SCID-hu mouse model is well established for studying VZV replication and determinants of tissue tropism (16); however, acquiring data has been hampered by having to euthanize the mice to measure virus growth. Sampling VZV-infected thymus-liver implants in the SCID-hu model has usually been done weekly or, at most, every 4 days. Moreover, the implants are different sizes, and so viral titers vary widely from animal to animal. To increase the sampling frequency and accuracy and, more importantly, to directly monitor VZV growth in live animals, we employed an IVIS, the SCID-hu thymus-liver mouse model, and VZVLuc to measure VZV spread in vivo each day for 1 week in the same mouse. The luciferase provides a visible marker for VZV in human tissues within living animals. Human fetal thymus and liver tissues were implanted under the left mouse kidney capsule. Two to three months later, the implants developed into a thymus-like organ consisting mainly of T cells. Viruses were grown in MeWo cells, and three SCID-hu mice with thymus-liver implants were inoculated by direct injection of a VZV-infected cell suspension. At different days postinoculation, the VZV replication in vivo was observed using an IVIS at 10 min after intraperitoneal injection of the SCID-hu mice with luciferin substrate. Bioluminescence was easily detected over the left flank of each of the three infected SCID-hu mice where the thymus-liver implants were located under the kidney capsule, demonstrating that the replication of VZVLuc can be monitored in living animals. Photon emissions from the infected implants were converted into a pseudocolored image (Fig. 2E). The color scale indicated regions of low emissions in purple and blue and high emissions in yellow and red. VZV was limited to the thymus-liver implants and did not transfer into the peritoneum or other organs, which was expected for this human-restricted virus. Notably, the average background emissions were similarly low for uninfected areas of inoculated mice (photon flux = 2,229 ± 510 photons/s/cm2/steradian) and over the kidney of a mock-inoculated control mouse (photon flux = 2,363 ± 334 photons/s/cm2/steradian). The boundaries of a region of interest were manually set to closely border the luminescent area in each mouse. Measurements of total photon flux were used as an indicator of VZV infection. This result demonstrates that VZVLuc spread can be monitored and quantified in vivo using an IVIS.
In vivo growth curve analysis of VZV.
VZV carrying a luciferase marker and the IVIS make measuring VZV replication in vivo possible. To measure VZV growth kinetics in vivo, three SCID-hu mice with human thymus-liver implants were inoculated with 4 × 103 PFU VZV-infected MeWo cells. These infected mice were scanned using IVIS imaging. Live imaging commenced within 4 h after inoculation, designated day zero, and then each mouse was imaged daily for 8 days. As shown in Fig. 3A, photon emissions from the infected implants increased daily until day 8 (only mouse C is shown). Each day, the bioluminescence signal from each mouse was quantified and plotted, and in vivo growth curves were generated (Fig. 3B). The kinetics of VZVLuc spread in three mice were similar and showed a general trend of exponential growth from day 1 to day 6 or 7, and then growth slowed or reached a steady state due to the fact that the viral infection within the limited implants had reached saturation (Fig. 3B). Based on these growth curves, VZV grew rapidly in human T cells in vivo, doubling approximately every 12 h, with maximal replication 7 days postinfection.
FIG. 3.
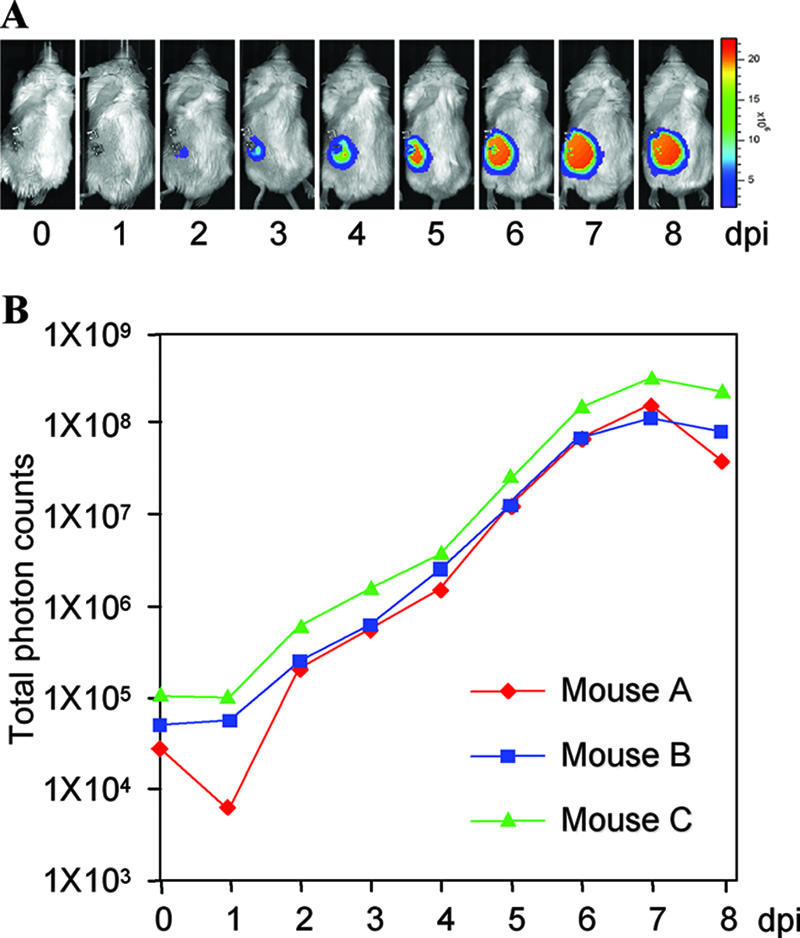
Measuring VZVLuc replication in SCID-hu mice. (A) Replication and progression of VZVLuc in human thymus-liver implants in SCID mice. Three SCID-hu mice implanted with thymus-liver implants were inoculated with VZVLuc. Using IVIS imaging, each mouse was scanned daily (from day 0 to day 8). Measurements were taken 10 min after intraperitoneal injection with d-luciferin substrate. Only images from mouse C are shown. Warmer colors indicate higher viral load; cooler colors indicate lower viral load. (B) VZV growth curves in vivo. Bioluminescence in three SCID-hu mice from the above experiment was measured, and VZV growth curves in human thymus-liver implants were generated.
VZVLuc contains a large DNA insert for the BAC vector (∼10 kb) and a luciferase expression cassette (∼3 kb). Even though it had been shown that VZV carrying the BAC sequence grows like its parental virus in vitro (Fig. 1D and Fig. 2D), it was not known whether it would grow normally in thymus-liver implants. Thus, VZVLuc was compared to the isogenic strain VZVLuc(-BAC), from which the BAC sequences were removed from its genome by Cre-lox recombination. Growth curve analysis in vivo indicated that removing the BAC sequences did not appear to confer a growth advantage to the VZVLuc strain since there were no major differences in photon emissions observed (data not shown). These results were representative of two separate experiments that used thymus-liver implants from different donors. From these data, we concluded that the presence of the BAC sequence did not interfere with VZV replication in T cells.
Generation of five deletion mutants of VZV.
To explore this novel luciferase VZV BAC system for studying VZV pathogenesis, five deletion mutants, from ORF0 (ORF0D) to ORF4 (ORF4D), were generated. To delete each of these ORFs from the VZV genome, a Kanr cassette was amplified by PCR using two primers containing 40-bp sequences homologous to the flanking sequences of each ORF. The PCR products were transformed into E. coli DY380 (32) harboring VZVLuc BAC and selected for chloramphenicol- and kanamycin-resistant colonies. In each transformant, the target ORF was replaced by the kanamycin cassette and deleted from its start to end sites (Fig. 4A). The correct deletion clones were checked first by comparing the HindIII digestion patterns with VZVLuc and ensuring that no abnormal deletions from the genomes were detected (Fig. 4B). Then the clones were confirmed by PCR analysis for the deletion of the target ORFs and the presence of adjacent ORFs (data not shown).
The mutants along with wild-type VZVLuc DNA were then transfected into MeWo cells to generate VZV deletion mutant viruses. For the clones with deletions of ORF1, ORF2, and ORF3 and parental VZVLuc, VZV plaques appeared between 3 and 5 days posttransfection, and recombinant virus grew like wild-type virus. The plaques from ORF0 deletion DNA developed significantly slower, resulting in smaller plaques. The ORF4 deletion DNA never produced any viruses after multiple transfections. These results suggested that for infection of MeWo cells, VZV ORF1, ORF2, and ORF3 are dispensable, ORF0 is required for optimal viral growth, and ORF4 is essential.
Analysis of five VZV deletion mutants.
Four VZV deletion mutant strains, ORF0D, ORF1D, ORF2D, and ORF3D, were amplified in culture, and titers were determined. Their growth properties in MeWo cells were compared with those of their parental VZVLuc strain. One hundred PFU of each mutant virus and wild-type virus was plated with fresh MeWo cells in 35-mm tissue culture wells in triplicate. Bioluminescence was recorded each day postinfection for 7 days using IVIS imaging after d-luciferin substrate was added to the wells for 10 min. Total photon counts from each well were measured, data from triplicates were averaged, and the growth curves of these viruses were generated. As shown in Fig. 4C, the ORF2D mutant grew as well as the wild type, and the ORF1D and ORF3D mutants grew slightly slower. In contrast, the ORF0D mutant showed a growth defect in MeWo cells, with slower kinetics and 10-fold less growth than wild-type and other mutant viruses.
The mutant viruses were tested in parallel with the VZVLuc strain in the SCID-hu model for their replication in human thymus-liver implants in vivo. Bioluminescence was measured at 4 h after inoculation and every day after for 7 days. For this rapid screening of in vivo phenotypes, three to five implants were inoculated with each virus. Most large implants produced strong and durable signals, while some small implants gave low or near-background signals. In addition, one mouse died on day 2. The poor signals due to small implants were removed from the data set, and the reliable numbers were averaged and used to generate in vivo growth curves of mice inoculated with different virus strains. As shown in Fig. 4D, mutant viruses ORF1D and ORF2D did not have a discernible growth defect in thymus-liver implants, and the ORF3D virus grew slightly, but not significantly, slower than the wild-type virus. Like wild-type VZV, these deletion mutants produced photon flux values of about 106 by 3 to 4 days after infection. In contrast, the implants inoculated with the ORF0D virus produced photon flux values that were approximately 100-fold (2 logs) less than those of VZVLuc or the other mutants. The rate of increase for ORF0D luminescence was lower, with the peak values obtained on day 5, and then levels declined. This defect of ORF0D in vivo is consistent with its defect in vitro, except that a more severe defect was observed.
Rescue of ORF0 and ORF4 growth defects.
To prove that the growth defects observed with the ORF0 and ORF4 mutants are due to the functions of the deleted genes rather than potential mutations in other regions of the genome, ORF0R and ORF4R viruses rescue were generated. ORF0 and ORF4 sequences were first amplified by PCR and cloned into pGEM-zeo-loxP. Both clones were confirmed by sequencing analysis. These wild-type ORF0 and ORF4 sequences were introduced back to ORF0D and ORF4D genomes, respectively, by homologous recombination. The rescued clones were confirmed by their antibiotic properties, HindIII digestions (Fig. 4B), and PCR analysis. ORF0R and ORF4R were then generated by transfection. Growth curve analysis using bioluminescence assays in vitro indicated that both ORF0R and ORF4R viruses fully recovered their growth defects of the deletion viruses and grew as well as the wild-type virus (Fig. 4E). These results confirm that ORF0 is required for optimal viral replication and that the ORF4 sequence is essential.
To further confirm the growth defect of ORF0D, and also to verify the correlation between the bioluminescence assay and the infectious center assay, 100 PFU of wild-type, ORF0 deletion (ORF0D), and ORF0D rescue (ORF0R) viruses were used to infect two sets of MeWo cells in triplets, as detailed in Materials and Methods. Comparison of the growth curves generated from both assays are shown in Fig. 4F. Both assays were correlated well for the growth of wild-type and ORF0R viruses, while for the ORF0D viruses, the data from the bioluminescence assay were slightly higher than those from the infectious center assay. This experiment has shown further that viral growth curves measured using both assays shared the same trend.
Monitoring VZVLuc replication in the implanted human skin in SCID mice.
VZVLuc was also tested for its spread and detection in human fetal skin xenografts in vivo. Human skin tissues were introduced into SCID mice as described previously (17). Four weeks after implantation, the VZVLuc strain and the ORF0D mutant and its rescue, ORF0R, viruses were inoculated in the skin tissues, and viral growth was monitored every 2 or 3 days for 15 days, using an IVIS. The wild-type and ORF0R viruses grew well and distinguishably from each other in the skin implants, whereas the ORF0D mutant grew significantly slower (Fig. 5).
FIG. 5.

Analysis of VZV ORF0D and ORF0R viruses in SCID mice with human skin implants. (A) The three panels show human fetal skin implants infected by VZVLuc and imaged at 15 dpi. ORF0D right (R), the right implant was infected with the ORF0 deletion mutant. The bioluminescence signal is significantly lower than that of its rescue virus. ORF0R left (L) and R, left and right implants were infected with the rescue virus of the ORF0 deletion mutant. High luciferase activities were detected in these implants. (B) Growth curve analysis of ORF0D (red circles), ORF0R (brown circles), and wild-type (WT, blue circles) viruses in the human skin tissues implanted in SCID mice. The implants without inoculation of VZV were used as a negative control (Mock, green triangles). Each curve was generated by averaging data from three implants. One set of data points is shown for a control of WT and Mock infection.
DISCUSSION
To date, knowledge of the molecular mechanisms of VZV pathogenesis remains relatively limited in comparison to that of other herpesviruses. Due to some unique features of this virus, such as its highly cell-associated nature in cell culture, genetic analysis based on conventional virological techniques, including plaque purification and high-multiplicity infections, becomes troublesome and challenging. To facilitate mutant virus generation, a VZV cosmid system derived from the pOka strain has been developed (6, 21). Cosmid-based mutagenesis has become a prevailing method to make genetically altered VZV recombinants. The entire genome of the pOka strain has been cloned as a VZV BAC previously (18) and in this study. Since cotransfection and homologous recombination after transfection are not required, recombinant VZVs can be produced efficiently.
Low-copy VZV BAC can be maintained in E. coli recA mutant strains (such as DY380 and DH10B) in a highly stable manner. The VZV BAC system allows for propagation of VZV DNA in E. coli, thus avoiding continual growth in cultured cells and preventing loss of VZV virulence. The VZV BAC system also allows easy DNA isolation from large volumes of bacterial culture. After multiple rounds of propagation and mutagenesis in E. coli, no detectable additional deletions or rearrangements of VZV genome were found. Sequencing analysis of ORF62, a large gene at ∼3.9 kb, indicated that no mutations were introduced into this ORF in the VZV BAC DNA. The growth kinetics indicated that the VZV virus generated from BAC DNA behaves indistinguishably from its parental pOka strain both in vitro and in vivo.
While the design of VZV BAC allows seamless removal of the 10-kb BAC vector sequence between two flanking loxP sites when VZV BAC DNA is cotransfected with a Cre expression plasmid into mammalian cells, growth curve analysis illustrated that VZV with and without the BAC vector have identical growth kinetics both in vitro and in vivo (Fig. 1D, Fig. 2D, and data not shown). This might be due to the fact that VZV has the smallest genome size compared with that of other human herpesviruses, and therefore its virion can tolerate extra DNA sequences in addition to its own genome without compromising viral replication. Furthermore, the GFP reporter gene embedded in this BAC vector and therefore future experimentation including plaque quantification and microscopy of viral infection are favorably enhanced and benefit from this visible feature of the VZV BAC virus replication and spread. If the BAC vector and the GFP have any interference with particular experiments, they can easily be removed from the viral genome by cotransfection of a Cre expression plasmid with VZV BAC DNA when VZV is produced.
The SCID-hu mouse model has been well developed to examine VZV pathogenesis and immunobiology in vivo (1, 3, 13, 33). If genetic changes are not essential for viral growth, VZV mutants can be further studied in SCID-hu mice with human thymus-liver and skin xenografts to illustrate the specific gene functions during VZV replication in fully differentiated human cells in vivo. However, acquiring quantitative data has been thwarted by the necessity of euthanizing the mice to measure viral titer. Sampling VZV-infected thymus-liver implants in the SCID-hu model has usually been done weekly or, at most, every 4 days. Moreover, VZV growth kinetics is rapid, and in order to monitor viral growth in vivo in a more accurate manner, more frequent sampling is required. Additionally, viral titers from one implant to another are quite different; a large number of SCID-hu mice are required to test each recombinant virus in order to obtain reliable data. Titration of VZV from the implants does not accurately reflect virus spread. Because the skin tissue is very tough, it cannot be dispersed to a single-cell suspension. Thymus-liver implants contain many dead T cells that were killed by VZV infection and are no longer infectious. In previous studies, the titer of VZV from infected implants (skin and thymus-liver) was not considered quantitative, and the kinetics of VZV replication was unclear. These factors have impeded efforts to study large numbers of VZV variants and have made it difficult to discern the minor phenotypic differences leading to pathogenesis.
To overcome the above-described obstacles, a luciferase expression cassette was inserted between ORF65 and ORF66 to achieve generation of the VZVLuc BAC. First, growth kinetic analyses showed that the presence of a luciferase gene did not interfere with VZV replication, both in vitro and in vivo. The growth kinetic analysis based on conventional infectious center assay and luciferase activity assays demonstrated that the intensities of bioluminescent signals correlate with virus titers (Fig. 2C and Fig. 4F). This enables subtle detection of viral replication as well as location over the course of infection in a real-time and noninvasive manner in human tissues. Moreover, in case a certain VZV variant bears a defect that alters the syncytium formation, the new luciferase activity approach will be superior to the conventional method because luciferase activities are indicators of viral loads instead of the number of infected cells. Therefore, the combination of VZVLuc and IVIS imaging not only makes monitoring VZV replication in live animals possible but also significantly reduces the number of SCID-hu mice required for experiments. Furthermore, this approach greatly increases experimental reproducibility and reliability. VZVLuc and IVIS imaging will be a very useful system with which to study the nature of VZV mutants. Although the bioluminescence assay has its advantages, this does not mean it could replace the infectious center assay. Rather, it serves as an easy alternative approach and a complement to the conventional assays. The nature of viral growth phenotypes found by bioluminescence assay should be further scrutinized using other methods.
VZV carries at least 70 unique ORFs, and many of their functions remain mysterious. Among those VZV ORFs, ORF0 (S/L), ORF1, ORF2, ORF13, ORF32, and ORF57 do not have homologs in herpes simplex virus (HSV) (8). This newly developed VZVLuc BAC clone can be efficiently altered by using the inducible lambda prophage system, providing homologous recombination in E. coli DY380 (32). The combination of VZV BAC technology, the SCID-hu model, the VZV genome carrying the luciferase gene, and live imaging forms a system with the potential capacity to generate and study much greater numbers of VZV strains than has been possible until now. In this paper, we reported the analysis of the small VZV genomic region, from ORF0 to ORF4, summarized in Table 1.
TABLE 1.
Summary of genetic analysis of VZV ORF0 to ORF4
| ORF | Location | Size (bp) | Gene product | HSV-1 orthologue | Mutant growth property
|
||
|---|---|---|---|---|---|---|---|
| In vitro | SCID-hu | Rescue | |||||
| 0 | 173-562 | 390 | Putative transmembrane protein | UL56 | Slow | Slow | Normal |
| 1 | 589-915a | 327 | Putative transmembrane protein | None | Normal | Normal | N/Ab |
| 2 | 1134-1850 | 717 | Unknown | None | Normal | Normal | N/A |
| 3 | 1908-2447a | 540 | Unknown | UL55 | Normal | Normal | N/A |
| 4 | 2783-4141a | 1,359 | Posttranscriptional regulator | UL54 | Essential | N/A | Normal |
Encoded on the bottom strand.
N/A, not applicable.
VZV ORF0, also known as ORFS/L, encodes a putative protein and has been suggested to play a role in altering cell adhesion molecules in infected cells (12). Intriguingly, we have observed a significant slow-growth phenotype in both cultured cells and SCID-hu T cells/skin xenografts, and the growth defect could be fully rescued when the wild-type copy was introduced back into its native site. Our results indicate that VZV ORF0 is a potential virulence gene, while the mechanism needs to be further investigated. It is possible that the deletion of ORF0 could interfere with end-of-genome functions rather than the function of the protein. This could be investigated by inserting stop codons in ORF0 rather than deleting the whole sequence.
VZV ORF1 encodes a putative membrane protein and has been shown to be dispensable for viral replication in vitro (7). VZV ORF2 encodes a phosphoprotein located in the membrane of infected cells and has been shown to be dispensable for viral replication in vitro and for establishment of latency in the cotton rat model (25). In this study, we have achieved similar results and, furthermore, have illustrated that VZV ORF1 and ORF2 are dispensable for viral replication in vivo. VZV ORF3 has not been studied before, while its HSV homolog, UL55, has been shown to be dispensable for viral replication and for establishment of latency (19). Our results have suggested that VZV ORF3 is dispensable for viral growth both in vitro and in vivo. VZV ORF4 encodes an immediate-early regulator that transactivates other viral genes in all three kinetic classes in a transient-expression system and enhances another immediate-early protein's, IE62's, transactivation activity. It has been shown that ORF4 is essential for viral replication and for establishment of latency in the cotton rat model (5, 24). Using the VZVLuc BAC system, a consistent result has been obtained in this study, serving as a control experiment for the system. Results from genetic analysis of ORF0 to ORF4 have also fully validated the use of the novel luciferase VZV BAC system to efficiently generate recombinant VZV variants and carry out subsequent viral growth kinetic analysis both in vitro and in vivo.
Future studies include functionally profiling the entire VZV genome, using this versatile and robust VZVLuc BAC system, identifying VZV tropism factors, and performing large-scale anti-VZV compound screening. Surely the principle could be expanded and applied to broader virological research.
Acknowledgments
We thank C. Patterson and A. Chu for critically reading the manuscript.
This work was supported by NIH grants AI050709 (H.Z.), AI052168 (J.F.M.), and AI053846 (A.M.A.).
Footnotes
Published ahead of print on 20 June 2007.
REFERENCES
- 1.Arvin, A. M. 2001. Varicella-zoster virus, p. 2731-2767. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Ball, K. D., and J. T. Trevors. 2002. Bacterial genomics: the use of DNA microarrays and bacterial artificial chromosomes. J. Microbiol. Methods 49:275-284. [DOI] [PubMed] [Google Scholar]
- 3.Besser, J., M. H. Sommer, L. Zerboni, C. P. Bagowski, H. Ito, J. Moffat, C.-C. Ku, and A. M. Arvin. 2003. Differentiation of varicella-zoster virus ORF47 protein kinase and IE62 protein binding domains and their contributions to replication in human skin xenografts in the SCID-hu mouse. J. Virol. 77:5964-5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boehncke, W. H. 1999. The SCID-hu xenogeneic transplantation model: complex but telling. Arch. Dermatol. Res. 291:367-373. [DOI] [PubMed] [Google Scholar]
- 5.Cohen, J. I., T. Krogmann, J. P. Ross, L. Pesnicak, and E. A. Prikhod'ko. 2005. Varicella-zoster virus ORF4 latency-associated protein is important for establishment of latency. J. Virol. 79:6969-6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen, J. I., and K. E. Seidel. 1993. Generation of varicella-zoster virus (VZV) and viral mutants from cosmid DNAs: VZV thymidylate synthetase is not essential for replication in vitro. Proc. Natl. Acad. Sci. USA 90:7376-7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen, J. I., and K. E. Seidel. 1995. Varicella-zoster virus open reading frame 1 encodes a membrane protein that is dispensable for growth of VZV in vitro. Virology 206:835-842. [DOI] [PubMed] [Google Scholar]
- 8.Cohen, J. I., and S. E. Straus. 2001. Varicella-zoster virus and its replication, p. 2707-2730. In D. M. Knipe and P. M. Howley (ed.), Fields virology, vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 9.Doyle, T. C., S. M. Burns, and C. H. Contag. 2004. In vivo bioluminescence imaging for integrated studies of infection. Cell Microbiol. 6:303-317. [DOI] [PubMed] [Google Scholar]
- 10.Gilden, D. H., B. K. Kleinschmidt-DeMasters, J. J. LaGuardia, R. Mahalingam, and R. J. Cohrs. 2000. Neurologic complications of the reactivation of varicella-zoster virus. N. Engl. J. Med. 342:635-645. [DOI] [PubMed] [Google Scholar]
- 11.Hambleton, S., and A. A. Gershon. 2005. Preventing varicella-zoster disease. Clin. Microbiol. Rev. 18:70-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kemble, G. W., P. Annunziato, O. Lungu, R. E. Winter, T.-A. Cha, S. J. Silverstein, and R. R. Spaete. 2000. Open reading frame S/L of varicella-zoster virus encodes a cytoplasmic protein expressed in infected cells. J. Virol. 74:11311-11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ku, C.-C., J. Besser, A. Abendroth, C. Grose, and A. M. Arvin. 2005. Varicella-zoster virus pathogenesis and immunobiology: new concepts emerging from investigations with the SCIDhu mouse model. J. Virol. 79:2651-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mallory, S., M. Sommer, and A. M. Arvin. 1997. Mutational analysis of the role of glycoprotein I in varicella-zoster virus replication and its effects on glycoprotein E conformation and trafficking. J. Virol. 71:8279-8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchini, A., H. Liu, and H. Zhu. 2001. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 75:1870-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moffat, J. F., and A. M. Arvin. 1999. Varicella-zoster virus infection of T cells and skin in the SCID-hu mouse model, p. 973-980. In M. A. Sande and O. Zak (ed.), Handbook of animal models of infection: experimental models in antimicrobial chemotherapy. Academic Press, San Diego, CA.
- 17.Moffat, J. F., M. D. Stein, H. Kaneshima, and A. M. Arvin. 1995. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J. Virol. 69:5236-5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagaike, K., Y. Mori, Y. Gomi, H. Yoshii, M. Takahashi, M. Wagner, U. Koszinowski, and K. Yamanishi. 2004. Cloning of the varicella-zoster virus genome as an infectious bacterial artificial chromosome in Escherichia coli. Vaccine 22:4069-4074. [DOI] [PubMed] [Google Scholar]
- 19.Nash, T. C., and J. G. Spivack. 1994. The UL55 and UL56 genes of herpes simplex virus type 1 are not required for viral replication, intraperitoneal virulence, or establishment of latency in mice. Virology 204:794-798. [DOI] [PubMed] [Google Scholar]
- 20.Netterwald, J., S. Yang, W. Wang, S. Ghanny, M. Cody, P. Soteropoulos, B. Tian, W. Dunn, F. Liu, and H. Zhu. 2005. Two gamma interferon-activated site-like elements in the human cytomegalovirus major immediate-early promoter/enhancer are important for viral replication. J. Virol. 79:5035-5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niizuma, T., L. Zerboni, M. H. Sommer, H. Ito, S. Hinchliffe, and A. M. Arvin. 2003. Construction of varicella-zoster virus recombinants from parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID-hu mouse model. J. Virol. 77:6062-6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rice, B. W., M. D. Cable, and M. B. Nelson. 2001. In vivo imaging of light-emitting probes. J. Biomed. Opt. 6:432-440. [DOI] [PubMed] [Google Scholar]
- 23.Sato, B., H. Ito, S. Hinchliffe, M. H. Sommer, L. Zerboni, and A. M. Arvin. 2003. Mutational analysis of open reading frames 62 and 71, encoding the varicella-zoster virus immediate-early transactivating protein, IE62, and effects on replication in vitro and in skin xenografts in the SCID-hu mouse in vivo. J. Virol. 77:5607-5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sato, B., M. Sommer, H. Ito, and A. M. Arvin. 2003. Requirement of varicella-zoster virus immediate-early 4 protein for viral replication. J. Virol. 77:12369-12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sato, H., L. Pesnicak, and J. I. Cohen. 2002. Varicella-zoster virus open reading frame 2 encodes a membrane phosphoprotein that is dispensable for viral replication and for establishment of latency. J. Virol. 76:3575-3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiraki, K., Y. Yoshida, Y. Asano, K. Yamanishi, and M. Takahashi. 2003. Pathogenetic tropism of varicella-zoster virus to primary human hepatocytes and attenuating tropism of Oka varicella vaccine strain to neonatal dermal fibroblasts. J. Infect. Dis. 188:1875-1877. [DOI] [PubMed] [Google Scholar]
- 27.Swaminathan, S., H. M. Ellis, L. S. Waters, D. Yu, E. C. Lee, D. L. Court, and S. K. Sharan. 2001. Rapid engineering of bacterial artificial chromosomes using oligonucleotides. Genesis 29:14-21. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi, M., T. Otsuka, Y. Okuno, Y. Asano, and T. Yazaki. 1974. Live vaccine used to prevent the spread of varicella in children in hospital. Lancet 2:1288-1290. [DOI] [PubMed] [Google Scholar]
- 29.Taylor, S. L., and J. F. Moffat. 2005. Replication of varicella-zoster virus in human skin organ culture. J. Virol. 79:11501-11506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Volpi, A. 2005. Varicella immunization and herpes zoster. Herpes 12:59. [PubMed] [Google Scholar]
- 31.Wang, W., C. E. Patterson, S. Yang, and H. Zhu. 2004. Coupling generation of cytomegalovirus deletion mutants and amplification of viral BAC clones. J. Virol. Methods 121:137-143. [DOI] [PubMed] [Google Scholar]
- 32.Yu, D., H. M. Ellis, E. C. Lee, N. A. Jenkins, N. G. Copeland, and D. L. Court. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 97:5978-5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zerboni, L., C. C. Ku, C. D. Jones, J. L. Zehnder, and A. M. Arvin. 2005. Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc. Natl. Acad. Sci. USA 102:6490-6495. [DOI] [PMC free article] [PubMed] [Google Scholar]